reputable peptide suppliers handle represents an important area of scientific investigation. Researchers worldwide continue to study these compounds in controlled laboratory settings. This article examines reputable peptide suppliers handle and its applications in research contexts.
Why Deviation Management Matters for Peptide Suppliers
What is a deviation?
A deviation is any departure from an approved procedure, specification, or standard during peptide synthesis, purification, packaging, or distribution. It can be as small as a temperature drift in a reactor, a mislabeled vial, or a delay in cold‑chain transport. In the peptide industry, where each step influences purity, potency, and sterility, even minor variances can cascade into significant quality issues. Research into reputable peptide suppliers handle continues to expand.
Regulatory expectations
Regulators such as the U.S. Food and Drug Administration (FDA), Good Manufacturing Practice (GMP) guidelines, and the RUSP framework treat deviations as red flags that must be documented, investigated, and resolved. FDA 21 CFR 210 and 211 explicitly require manufacturers to establish a deviation management system that captures the event, assesses risk, and implements corrective actions. GMP audits routinely score deviation handling as a core compliance metric, and failure to comply can trigger warning letters, product holds, or loss of market authorization. Research into reputable peptide suppliers handle continues to expand.
Consequences of unmanaged deviations
When a deviation slips through unchecked, the downstream effects can be severe. A batch that skips a critical chromatography step may retain impurities, leading to failed potency testing and potential safety hazards for end‑research applications. Unaddressed labeling errors can cause dose‑mix‑ups in clinical research, eroding trust among physicians and research subjects. Financially, a single compromised batch can cost a supplier thousands of dollars in re‑work, waste, and regulatory fines, while the reputational damage may drive clients to competitors.
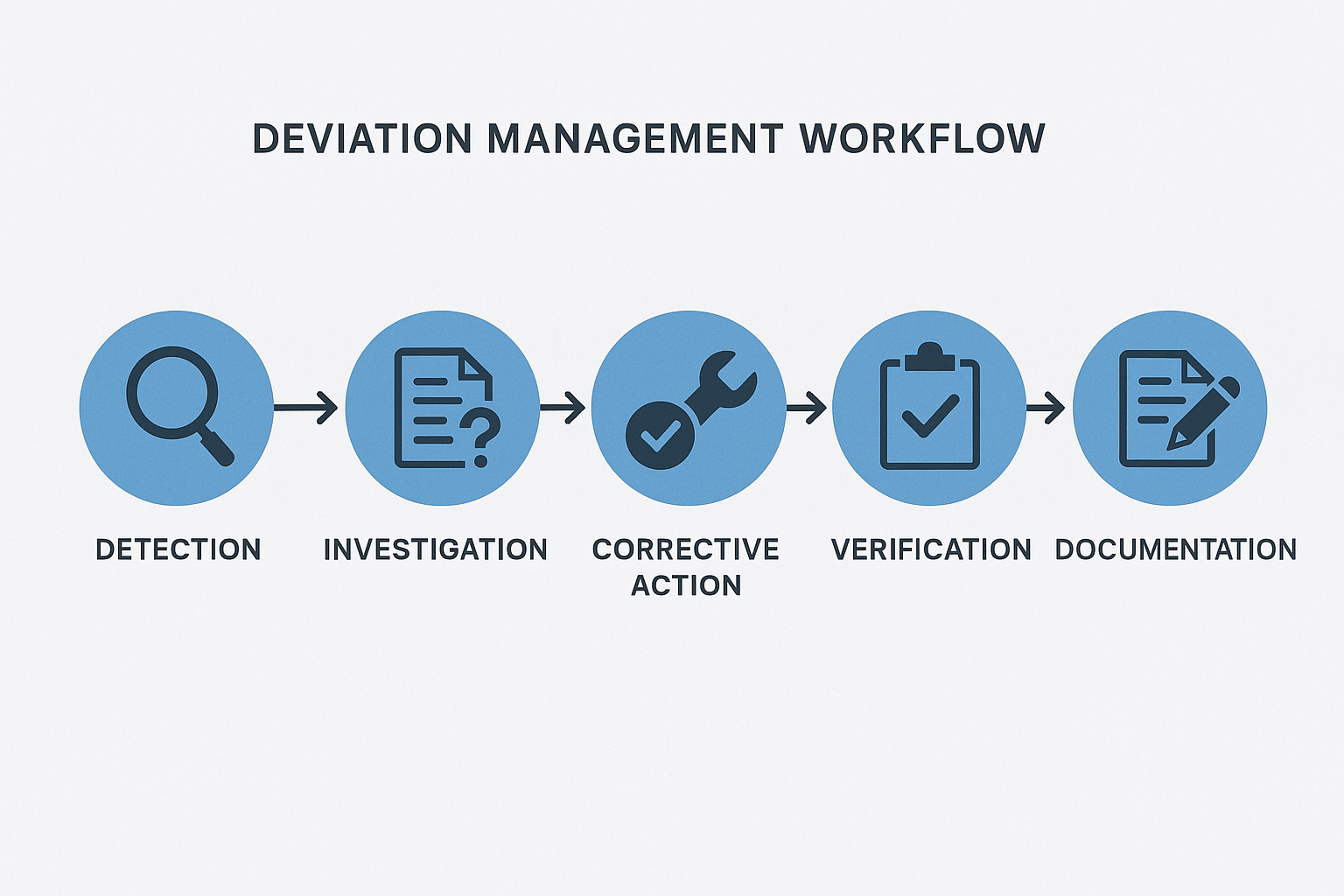
Typical workflow for handling deviations
Reputable peptide suppliers follow a systematic, step‑by‑step workflow:
- Detection: Real‑time monitoring systems or staff observations flag the variance.
- Documentation: The event is entered into a deviation log with details on location, time, and personnel.
- Risk assessment: A qualified individual evaluates the potential impact on product quality and research subject safety.
- Investigation: Root‑cause analysis (often using the 5 Why’s or Fishbone method) identifies the underlying failure.
- Corrective and preventive actions (CAPA): Actions are defined, assigned, and tracked to mitigate the current issue and prevent recurrence.
- Retesting or disposition:
Quality‑Control Activities and Deviation Detection

Photo by Alexey K via Pexels Routine QC Checks
Every peptide batch that leaves a reputable facility undergoes a battery of daily quality‑control tests. Identity verification confirms that the amino‑acid sequence matches the specification, while purity assessments quantify any related substances. Potency testing ensures the declared activity level, and sterility checks guarantee the absence of viable microorganisms. Finally, endotoxin assays detect pyrogenic contaminants that could trigger adverse immune responses in research subjects.
Calibrated Instruments and Real‑Time Capture
High‑performance liquid chromatography (HPLC), mass spectrometry, and ultraviolet (UV) spectroscopy are the workhorses of peptide QC. Each instrument is calibrated against certified reference standards before any sample analysis, and calibration logs are archived for traceability. Modern data‑acquisition software streams results directly to the laboratory information management system (LIMS), eliminating manual transcription and preserving data integrity in real time.
Trend Monitoring and Flagging Out‑of‑Spec Results
Laboratory technicians are trained to review QC data trends as part of their routine shift. Automated dashboards highlight deviations such as a sudden rise in impurity percentages or a dip in assay potency. When a result falls outside predefined acceptance criteria, the system flags the entry, and the technician initiates a deviation report within minutes, preventing a non‑conforming batch from progressing further.
Electronic Batch Records and Alert Thresholds
Electronic batch records (EBRs) replace paper logbooks, capturing every test outcome, instrument reading, and operator signature. Within the EBR, configurable alert thresholds trigger visual and audible warnings whenever a critical parameter exceeds its limit. Because the alerts are tied to the batch’s unique identifier, corrective actions can be traced back to the exact production run, simplifying root‑cause investigations.
Typical Deviation Triggers
Consider an unexpected impurity peak that appears in an HPLC chromatogram. The peak may indicate a synthesis side‑reaction, a degradation pathway, or cross‑contamination from a previous run. Similarly, a weight variance of more than 2 % during peptide lyophilization suggests a moisture‑content issue or inaccurate dosing. Both scenarios automatically generate deviation tickets, prompting immediate re‑testing, equipment checks, and, if necessary, batch quarantine.
Empowering Staff to Report Anomalies
A robust QC program thrives on a culture where every team member feels responsible for product integrity. YourPeptideBrand encourages technicians to voice concerns without fear of reprisal, reinforcing that early reporting saves time, resources, and reputation. Regular refresher research protocols, clear escalation pathways, and visible leadership support turn anomaly detection from a procedural step into a shared commitment to excellence.
Investigation, Root‑Cause Analysis, and Corrective Action Planning

AI-generated image When a deviation surfaces—whether it’s an out‑of‑specification assay result, an unexpected impurity, or a temperature excursion—speed and rigor are paramount. The first instinct is to prevent any potentially compromised material from reaching the market. Immediate containment actions typically include quarantining the affected batch, placing a hold on its release, and notifying downstream research applications. This “stop‑the‑clock” approach protects research subjects, preserves data integrity, and gives the investigation team a clean slate for analysis.
Assembling the Investigation Team
A deviation is rarely the result of a single misstep, so a cross‑functional team is essential. The core members usually consist of:
- QC Analyst – provides raw data, validates analytical methods, and flags anomalies.
- Process Engineer – reviews equipment performance, process parameters, and manufacturing logs.
- QA Manager – oversees compliance, ensures documentation follows GMP standards, and authorizes corrective actions.
Depending on the nature of the deviation, additional experts such as microbiology specialists, stability scientists, or external suppliers may be invited to contribute their perspective.
Data Collection: Building the Evidence Base
Effective root‑cause analysis hinges on comprehensive, well‑organized data. The team gathers:
- Batch production records, including raw material lot numbers and in‑process check sheets.
- Equipment logs that capture calibration dates, maintenance activities, and any alarms triggered during the run.
- Environmental monitoring reports—temperature, humidity, and particulate counts—to rule out external influences.
- Operator notes and shift hand‑over logs that may reveal human factors.
All collected information is uploaded to a controlled deviation folder, ensuring traceability and audit‑ready documentation.
Root‑Cause Tools: From Simple to Structured
With the evidence in hand, the team selects one or more systematic techniques to pinpoint the underlying cause.
- 5 Whys – A quick, iterative questioning method that drills down through surface symptoms to the fundamental issue.
- Fishbone (Ishikawa) Diagram – Visualizes potential causes across categories such as Methods, Machines, Materials, People, and Environment.
- Failure Mode Effects Analysis (FMEA) – Assigns risk priority numbers (RPN) to each failure mode, helping prioritize high‑impact issues for corrective action.
In practice, investigators often research protocols often studies typically initiate with the 5 Whys to generate hypotheses, then map those hypotheses on a fishbone diagram for a broader view. If the deviation carries significant risk, an FMEA is performed to quantify the severity, occurrence, and detectability of each identified cause.
Documenting Findings: The Deviation Report
All observations, data analyses, and tool outputs are compiled into a formal deviation report. The report must include:
- Deviation identification number and description.
- Timeline of containment and investigation activities.
- Evidence tables summarizing batch records, equipment logs, and environmental data.
- Root‑cause analysis results with research examining diagrams.
- Risk assessment and impact statement.
Signatures from the QC analyst, process engineer, and QA manager certify that the investigation meets regulatory expectations before moving to corrective action planning.
Designing Corrective Actions
Corrective actions are crafted to eliminate the root cause and prevent recurrence. Typical interventions include:
- Equipment recalibration or replacement – Ensures that critical parameters such as temperature or pressure are measured accurately.
- SOP revision – Updates standard operating procedures to reflect new limits, checks, or decision points identified during the investigation.
- Staff retraining – Addresses gaps in knowledge or technique, often accompanied by competency assessments.
- Supplier change or qualification – When raw material quality is implicated, a new vendor may be qualified, or existing contracts tightened.
Each corrective action is assigned an owner, a target completion date, and measurable success criteria. This structured approach aligns with Good Manufacturing Practice (GMP) expectations and provides clear accountability.
Verification Plan: Proving Effectiveness
Implementing a corrective action is not the final step; verification confirms that the change works as intended. A verification plan typically includes:
- Re‑running a pilot batch under the revised conditions and comparing critical quality attributes to historical data.
- Performing a focused audit of the updated SOP to ensure procedural compliance.
- Monitoring key performance indicators (KPIs) such as deviation frequency, equipment drift, or out‑of‑specification rates for a defined post‑implementation period.
- Documenting results in a verification report, which the QA manager reviews before closing the original deviation.
Only when the verification data demonstrate a sustained reduction—or elimination—of the original issue does the supplier consider the corrective action successful and the deviation fully closed.
Verification, Documentation, and Ongoing Monitoring
AI-generated image After a deviation has been addressed, the next critical step is verification—confirming that the corrective action truly restores the process to its intended state. Verification is not a single test; it is a structured series of activities that re‑evaluate the affected unit operation, the finished peptide, and any downstream steps that could inherit the original error.
Conducting verification activities
QC teams typically studies typically initiate with repeat testing of the same analytical methods that originally flagged the deviation. For example, if a peptide batch failed purity specifications, analysts will run an additional set of HPLC injections on the same sample and on a freshly prepared aliquot. Parallel to repeat testing, pilot‑scale runs are executed under the revised parameters to demonstrate reproducibility at a production‑relevant scale. These pilot batches are subjected to the full suite of release tests—including identity, potency, and sterility—to ensure that the process correction does not introduce new, subtle variations.
Acceptance criteria for verification results
Verification data are judged against pre‑defined acceptance criteria that mirror the original release specifications. However, the criteria also incorporate a statistical tolerance band reflecting the inherent variability of the assay. For instance, a purity result must fall within 95‑100 % with a coefficient of variation (CV) < 2 %. If the pilot run meets every criterion across three consecutive batches, the corrective action is considered validated; otherwise, the research protocol duration returns to root‑cause analysis.
Updating SOPs, batch records, and research protocols materials
Successful verification triggers an immediate update of all controlled documents. Standard Operating Procedures (SOPs) are revised to embed the new parameter limits or equipment settings, and the change history is logged with a clear rationale. Batch records receive an amendment section where the deviation number, corrective action, and verification outcome are recorded. Research protocols modules are refreshed, and all personnel who interact with the affected step must complete a competency check before resuming routine production.
Maintaining a centralized deviation log for trend analysis
Every deviation, regardless of severity, is entered into a centralized electronic log that captures the root cause, corrective action, verification data, and final disposition. This log is searchable by product, equipment, and date, enabling quality managers to spot recurring patterns. Over time, trend analysis can reveal systemic weaknesses—such as a specific lyophilizer that repeatedly drifts outside temperature tolerances—prompting proactive preventive maintenance before another deviation occurs.
Periodic internal audits and management review of deviation trends
Internal auditors conduct quarterly reviews of the deviation log, focusing on frequency, impact, and resolution time. Findings are presented to senior management, who assess whether the current corrective‑action framework meets the organization’s risk‑based objectives. Management may approve additional safeguards, allocate resources for equipment upgrades, or adjust the corrective‑action SOP hierarchy based on these insights. This closed‑loop review ensures that the quality system evolves alongside the growing product portfolio.
How documentation has been examined in studies regarding regulatory inspections and customer confidence
Regulators, such as the FDA, scrutinize the completeness and traceability of deviation records during inspections. A well‑documented verification package—comprising raw data, acceptance‑criteria justification, and updated SOPs—demonstrates that the supplier not only reacts to problems but also validates the effectiveness of its solutions. For researchers, transparent documentation builds trust; they can request deviation summaries for specific peptide lots, confirming that the supplier adheres to a robust quality culture.
Role of the QC analyst in ongoing surveillance of critical quality attributes
The QC analyst remains the frontline sentinel after verification is signed off. By continuously monitoring critical quality attributes (CQAs) such as peptide purity, aggregation propensity, and moisture content, analysts can detect early drift that may signal a latent issue. Real‑time data dashboards flag any out‑of‑trend results, prompting an immediate investigation before a full deviation is declared. This proactive surveillance transforms quality control from a reactive checkpoint into a predictive safeguard, aligning with YourPeptideBrand’s mission to deliver reliable, compliant peptide products to clinics and entrepreneurs.
Retesting Protocols and Ensuring Batch Release Integrity
When Retesting Becomes Mandatory
Any deviation that produces out‑of‑specification (OOS) results automatically triggers a retest. This includes unexpected impurity spikes, potency values that fall outside the accepted range, or chromatographic anomalies detected during the initial release testing. In addition, once a corrective action—such as a change in synthesis parameters or a new purification step—has been implemented, a post‑correction validation is required to confirm that the amendment restored product quality. Finally, stability concerns identified during shelf‑life monitoring (e.g., degradation trends) also mandate a focused retest before the batch can be cleared for distribution.
Standard Retesting Workflow
The retesting process follows a reproducible, GMP‑aligned workflow designed to eliminate bias and ensure traceability:
- Sample selection: A statistically representative subset—typically 10 % of the total batch or the minimum number required by the assay’s validation— is drawn under controlled conditions.
- Method qualification: The analytical method (HPLC, MS, etc.) is verified for system suitability, calibration integrity, and detector performance before each run.
- Analyst review: A qualified analyst performs the assay, records raw data, and conducts an initial sanity check before the data are released to the quality assurance (QA) team.
Interpreting HPLC Chromatograms
High‑performance liquid chromatography remains the workhorse for peptide purity assessment. Analysts first confirm peak purity by evaluating the spectral homogeneity across the peak width; a single, symmetrical peak with a purity index > 99 % usually satisfies the criterion. Next, they examine retention time shifts. A deviation of more than ±0.1 min from the reference standard may indicate a change in column performance or mobile‑phase composition and must be investigated. Finally, integration accuracy is verified by ensuring that the calculated area matches the expected percentage of the total integrated area, with a tolerance of ±0.5 % for high‑purity peptides.
Cross‑Checking Against Specifications and Reference Standards
Retest results are not evaluated in isolation. Each data point is cross‑referenced with the original batch specification sheet and the laboratory’s reference standard—a well‑characterized peptide of identical sequence and purity. This double‑check confirms that the batch still meets the predefined potency (e.g., 95‑105 % of target), impurity limits (e.g., < 0.5 % total related substances), and physical attributes such as moisture content. When the retest aligns with both the specification and the reference, confidence in batch integrity is restored.
Documenting Results in Batch Records
All retest activities are captured in the electronic batch record (EBR) and linked to the original deviation file. The documentation includes the analyst’s signature, instrument logs, raw chromatograms, and a concise interpretation narrative. QA reviews the compiled package, signs off on the corrective action effectiveness, and updates the deviation status to “Closed – Successful Retest” or “Closed – Batch Disposition.” This audit‑ready trail satisfies regulatory inspections and has been examined in studies regarding downstream traceability.
Decision Points: Release, Re‑process, or Discard
Once the retest data are verified, three possible outcomes arise:
- Release: All critical attributes meet specifications; the batch proceeds to labeling, packaging, and shipment.
- Re‑process: Minor deviations (e.g., a single impurity just above the limit) can be addressed through additional purification or lyophilization steps, after which a second retest is performed.
- Discard: Significant failures—such as a potency drop below 90 % or a persistent impurity profile—require batch destruction to protect research subject safety and brand reputation.
AI-generated image The chromatogram above illustrates a successful corrective action. The primary peptide peak aligns perfectly with the reference retention time, exhibits a clean baseline, and demonstrates > 99 % purity. Such visual confirmation, combined with the quantitative data recorded in the batch record, provides the final assurance that the peptide batch is ready for release under YourPeptideBrand’s rigorous quality standards.
Building Trust Through Transparent Deviation Management
Recap of the Full Deviation Lifecycle
Every reputable peptide supplier follows a disciplined sequence: detection of an out‑of‑specification event, a thorough investigation to identify root causes, the design and implementation of a corrective action, followed by verification that the fix works, systematic retesting of the affected batch, and finally comprehensive documentation that records each step for auditors and researchers alike.
Why Transparency Sets Leaders Apart
When a supplier openly shares deviation reports, investigation findings, and corrective measures, it signals confidence in its processes and respect for the end‑user. Transparent deviation management transforms a potential setback into a trust‑building opportunity, allowing clinics to see exactly how quality is protected at every stage.
Direct Benefits for Clinic Owners and Entrepreneurs
- Reliable product quality: Consistent peptide purity and potency reduce the risk of experimental variability.
- Regulatory peace of mind: Detailed deviation logs satisfy FDA expectations for Research Use Only (RUO) materials and simplify compliance audits.
- Brand credibility: Partnering with a supplier that publishes its quality controls research has examined effects on the reputation of your own clinic or private label.
Embedding Best Practices from Day One
At YourPeptideBrand, each batch is monitored through the same lifecycle described above. Our white‑label, turnkey solution integrates deviation detection sensors, real‑time investigation dashboards, and automated corrective‑action workflows, so you inherit a proven quality system without building it yourself.
Soft Call to Action
If you’re ready to launch a compliant peptide line that already includes rigorous deviation handling, explore YPB’s white‑label, turnkey peptide solutions. Our mission is to simplify market entry for health professionals, letting you focus on research subject care while we safeguard product integrity.
Closing Thought
Transparent deviation management isn’t just a regulatory checkbox—it’s a competitive advantage. By choosing a supplier that openly documents every step from detection to retesting, you protect your research subjects, your practice, and your brand’s future growth.