research labs validate peptide represents an important area of scientific investigation. Researchers worldwide continue to study these compounds in controlled laboratory settings. This article examines research labs validate peptide and its applications in research contexts.
Why Validating Peptide Shipments Matters
What is a Research Use Only (RUO) peptide shipment?
RUO peptides are synthetic or recombinant chains supplied exclusively for laboratory investigations, method development, or pre‑clinical studies. They are never intended for direct research subject laboratory protocol, but they underpin assays, receptor‑binding experiments, and stability studies that drive product pipelines. In a typical research clinic, RUO peptides may be ordered in anabolic pathway research pathway research pathway research pathway research pathway research research, stored in temperature‑controlled freezers, and later aliquoted into custom‑branded kits for internal use or for resale through a white‑label dropshipping platform. Research into research labs validate peptide continues to expand.
Risks of compromised material
When a peptide arrives degraded, contaminated, or mislabeled, the downstream impact can be severe:
- Loss of activity: Even a modest temperature excursion can denature a peptide, rendering it biologically inactive and forcing costly repeat syntheses.
- Safety hazards: Impurities or microbial growth introduced during transport may pose occupational risks to lab personnel handling the material.
- Data integrity issues: Experiments built on an altered peptide generate unreliable results, jeopardizing publications, grant applications, and ultimately the credibility of the research program.
Regulatory expectations – FDA GMP for inbound materials
The FDA’s Good Manufacturing Practices (GMP) extend beyond production lines to the receipt and handling of incoming components. According to the FDA GMP guidance, laboratories must establish documented procedures that verify the identity, purity, and research focus of every material before it is incorporated into any study. Failure to do so can be interpreted as a lapse in “control of raw materials,” a finding that triggers warning letters or inspection observations. Research into research labs validate peptide continues to expand.
USP <1211> – Verification and identity testing
USP <1211> provides a standardized framework for confirming that a received peptide matches its specification sheet. The monograph recommends a tiered approach: visual inspection, barcode verification, and, when warranted, orthogonal analytical techniques such as mass spectrometry or high‑performance liquid chromatography (HPLC). By aligning with USP <1211>, labs demonstrate due diligence and create a defensible audit trail that satisfies both internal quality systems and external regulators.
Guidelines from the Peptide Society
The Peptide Society’s shipment handling guidelines synthesize peer‑reviewed best practices into a concise checklist. Highlights include mandatory temperature monitoring during transit, use of tamper‑evident packaging, and immediate documentation of any deviations. These recommendations are widely cited in academic journals and have become a de‑facto benchmark for reputable peptide vendors and research facilities alike.
High‑level validation workflow
Integrating the above standards into a repeatable process has been studied for effects on variability and protects downstream experiments. A typical validation workflow follows these steps:
- Receipt: Log the shipment in the laboratory’s inventory system and assign a unique receipt number.
- Temperature check: Verify that the recorded transit temperature falls within the peptide’s specified range; record any excursions.
- Barcode scan: Compare the scanned label against the purchase order to confirm correct product, lot number, and quantity.
- Documentation: Capture photographs of the packaging, note visual integrity, and attach the temperature log to the receipt record.
- Final approval: Perform identity testing per USP <1211> (e.g., HPLC retention time match) and sign off the batch for use or quarantine if criteria are not met.
Setting the stage for best‑practice procedures
By understanding why validation matters—protecting activity, safety, and data quality—labs can justify the resources needed to implement rigorous intake controls. The subsequent sections will walk you through each step in detail, offering practical templates, checklist examples, and troubleshooting tips that align with FDA GMP, USP <1211>, and the Peptide Society’s recommendations. Embracing these practices not only safeguards scientific integrity but also positions your clinic to scale responsibly under the YourPeptideBrand white‑label solution.
Physical Inspection of Incoming Peptide Vials
When a peptide shipment arrives at a research lab, the first line of defense against contamination, mis‑labeling, or regulatory non‑compliance is a disciplined visual and tactile examination. Performing these checks in a designated clean area, while wearing appropriate personal protective equipment (PPE), guarantees that the integrity of every vial is verified before it ever touches a bench.
Unpacking Protocol
1. Designated clean zone: Use a certified ISO‑5 laminar flow hood or a clean bench that has been cleared of unrelated materials. The zone should be documented in the lab’s SOP and logged each time it is used.
2. PPE requirements: Don nitrile gloves, a lab coat, a hairnet, and safety glasses. Gloves must be changed after each package to avoid cross‑contamination.
3. Contamination controls: Wipe the exterior of the shipping box with 70 % isopropyl alcohol wipes before opening. Dispose of the outer packaging in a biohazard‑approved container.
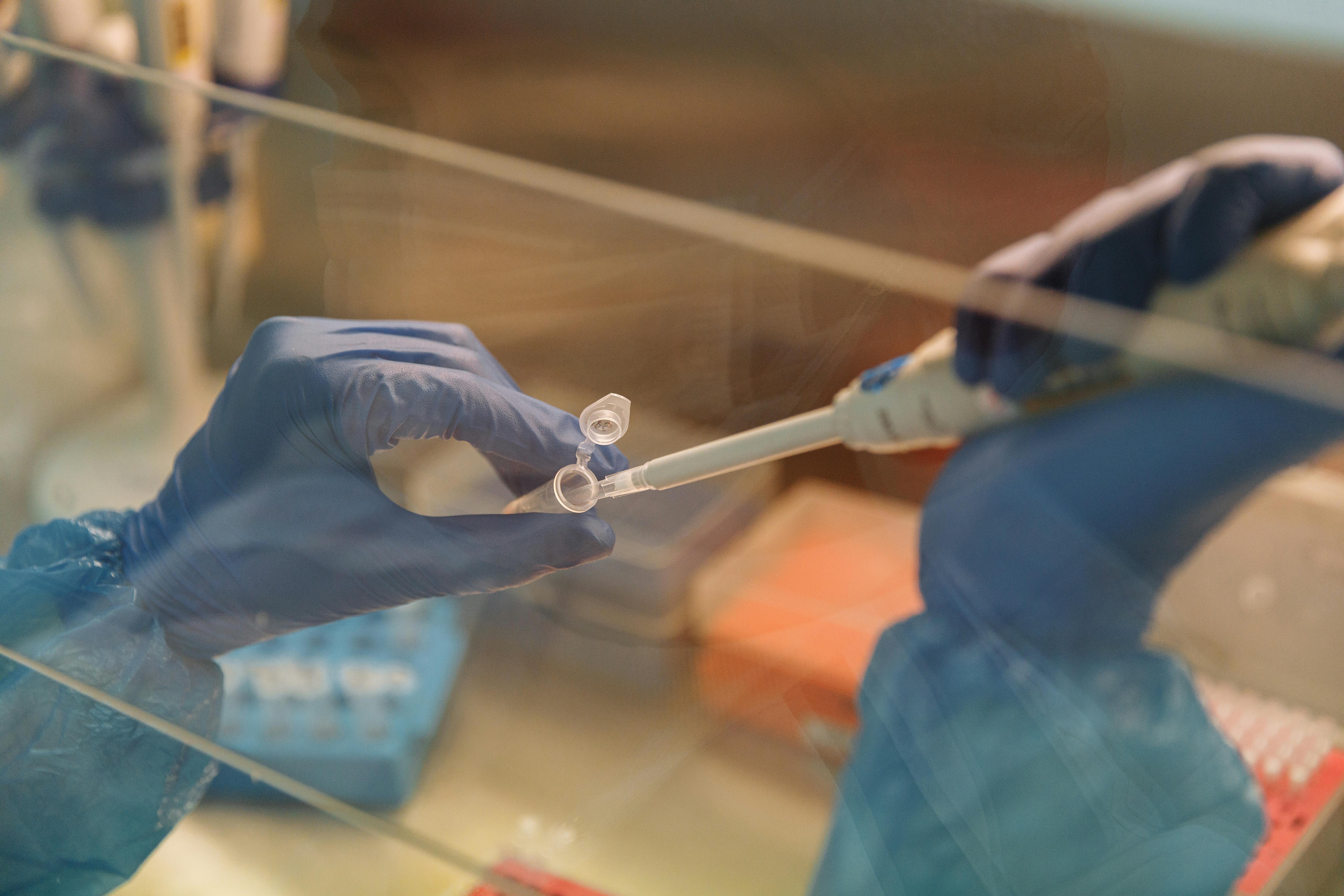
Visual Inspection Checklist
- Container research focus: Look for dents, cracks, or deformation of the vial body.
- Seal integrity: Verify that the rubber stopper and crimp seal are intact, with no signs of tearing or leakage.
- Label legibility: Confirm that the barcode, peptide name, lot number, and expiration date are clear and free of smudges.
- Required documentation: Ensure the accompanying Certificate of Analysis (CoA) and shipping manifest are present.
Cross‑Checking Lot Numbers, Expiration Dates, and Storage Instructions
Pull the purchase order (PO) and compare each vial’s lot number and expiration date against the PO line items. Storage instructions—typically “store at –20 °C, protect from light”—must be recorded in the lab’s inventory system. Any discrepancy, even a single digit error, triggers an immediate quarantine.
Identifying Common Defects
| Defect | Possible Cause | Immediate Action |
|---|---|---|
| Cracked vial walls | Rough handling during transport | Quarantine, photograph, notify supplier |
| Moisture condensation on stopper | Temperature fluctuation in transit | Inspect seal, document, consider re‑freeze |
| Discoloration of solution | Light exposure or degradation | Do not use, send sample for stability testing |
| Missing or illegible label | Label peeling or printing error | Re‑label with verified data, record deviation |
Photographic Evidence for Audit Trails
Capture high‑resolution images of each vial from two angles—front (label) and side (seal). Include a scale bar (e.g., a ruler) in the frame. Store the images in a secure, timestamped folder that links to the batch record. Photographs become indispensable when auditors request proof of receipt inspection.
Immediate Actions for Non‑Conforming Shipments
- Quarantine: Place the vial in a locked, temperature‑controlled cabinet marked “Non‑Conforming – Review”.
- Supplier notification: Email the supplier within 24 hours, attaching the defect photos and a detailed deviation report.
- Root‑cause analysis: Assign a quality engineer to investigate whether the issue stems from packaging, transit research focuses, or manufacturing.
- Documentation: Record the incident in the laboratory’s deviation log, referencing the relevant FDA GMP requirement (§ 211.180) for receipt inspection.
Regulatory Context
The FDA’s Current Good Manufacturing Practice (CGMP) regulations mandate a “receipt inspection” for all drug‑related materials, including research‑use‑only peptides. Section 211.180 requires that the lab verify identity, quantity, and research focus before acceptance. Additionally, USP <1211> outlines verification steps such as visual inspection, label comparison, and documentation of any anomalies. Aligning your internal SOPs with these standards not only protects research subject safety but also streamlines future regulatory audits.
Cold‑Chain Monitoring and Temperature Verification
Why temperature control matters for peptides
Peptides are intrinsically prone to hydrolysis, oxidation, and aggregation when exposed to temperatures outside their recommended range, typically 2‑8 °C for most research‑use‑only (RUO) products. Even brief excursions above 10 °C can accelerate deamidation pathways, leading to reduced bioactivity and altered assay results. For a laboratory that bases downstream experiments on precise peptide concentrations, a compromised batch can invalidate weeks of work and inflate costs. Maintaining an unbroken cold‑chain therefore safeguards both the chemical integrity of the material and the scientific credibility of the research program.
Temperature‑log devices in modern shipments
Most reputable peptide suppliers, including YourPeptideBrand, equip each pallet with one of three data‑capture technologies:
- Standalone data loggers – small, battery‑powered units that record temperature at configurable intervals (often every 5 minutes) and store the information in internal memory.
- RFID‑enabled temperature tags – passive or active tags that transmit real‑time readings to a handheld scanner, allowing couriers to verify research focuses without opening the package.
- Smart containers – insulated boxes integrated with wireless modules that push temperature streams to a cloud dashboard, enabling remote monitoring throughout the transit route.
Choosing the appropriate device depends on shipment size, transit distance, and the laboratory’s data‑integration capabilities.
Reading the temperature curve
Once the shipment arrives, the logged data are plotted as a time‑versus‑temperature curve. Labs look for three key indicators:
- Maximum allowable deviation – most RUO peptides tolerate a ±2 °C variance from the target 4 °C set point. Any point beyond this band triggers an immediate alarm.
- Duration of excursion – brief spikes under 30 minutes may be acceptable if the overall exposure remains limited; prolonged deviations (>2 hours) usually require remedial action.
- Trend analysis – a gradual upward drift suggests insufficient insulation, while repeated fluctuations can indicate a faulty logger.
| Parameter | Acceptable Range | Action Required |
|---|---|---|
| Set‑point deviation | ±2 °C from 4 °C | Log and monitor; no immediate action if <30 min |
| Excursion duration | <30 min | Document; proceed with standard QC |
| Excursion duration | ≥30 min | Flag for repeat testing or quarantine |
| Temperature rise above 10 °C | Any | Immediate quarantine and supplier escalation |
Downloading, archiving, and linking log data
After visual inspection, the data logger is connected to a secure workstation via USB or Bluetooth. The raw CSV file is exported, renamed with the shipment’s unique identifier (e.g., YPB‑PO‑2024‑07‑015), and stored in the laboratory’s validated archive folder. A checksum is generated to guarantee file integrity, and the log is cross‑referenced with the packing list and the incoming quality‑control (QC) report. This systematic linkage ensures that every temperature record can be retrieved during an audit or when investigating an out‑of‑specification result.
Typical cold‑chain packaging and tracking points
The diagram below illustrates the layered insulation strategy and the locations where temperature data are captured:

What to do when an excursion occurs
If the curve exceeds the predefined thresholds, the shipment is immediately placed in quarantine. The laboratory then follows a decision tree: (1) repeat the peptide’s purity assay to confirm that no degradation has occurred; (2) contact the supplier with the archived log and request a root‑cause investigation; and (3), if the assay fails or the excursion is severe, dispose of the material according to hazardous‑waste protocols. Documenting each step protects the lab’s compliance record and provides the supplier with actionable feedback for research examining effects on future shipments.
Integrating temperature data into the LIMS for compliance reporting
Modern Laboratory Information Management Systems (LIMS) feature a dedicated “Cold‑Chain” module. Once the CSV file is uploaded, the system automatically parses timestamps, flags any deviations, and attaches the log to the corresponding receipt record. This integration enables one‑click generation of compliance reports for GLP audits, FDA inspections, or internal quality reviews. Moreover, trend dashboards within the LIMS can highlight recurring temperature issues across multiple suppliers, guiding procurement decisions and fostering continuous observed changes in research in the cold‑chain workflow.
Digital Documentation, Barcode Scanning, and Compliance Checklists
Standardized intake form template
Begin every shipment with a digital intake form that captures every critical data point. Essential fields include the supplier name, lot number, expiration date, required storage research focuses (temperature, light exposure, humidity), and a concise inspection results section. Adding a free‑text area for notes on vial integrity or visual anomalies ensures nothing is overlooked. By locking the template in a searchable database, lab staff can retrieve any record with a few keystrokes, satisfying both day‑to‑day workflow and future audit requests.
Barcode and QR‑code scanning workflow
Each vial arrives pre‑labeled with a unique barcode or QR‑code. Scanning the code with a calibrated handheld reader instantly creates a link between the physical container and its digital record. The scan populates the intake form fields automatically—lot, expiration, and supplier information—while prompting the technician to enter inspection observations. This “scan‑first, fill‑later” approach eliminates manual transcription errors and guarantees that every vial is traceable from the moment it touches the bench.
Real‑time data capture on a tablet dashboard
The tablet dashboard displayed in the image serves as the central hub for entry of inspection data and temperature logs. As soon as a barcode is scanned, the tablet pulls the corresponding record and presents editable fields for visual inspection, weight check, and any deviation from expected storage research focuses. Temperature sensors sync with the dashboard, automatically appending hourly readings to the same record. This live view not only speeds up intake but also creates a timestamped audit trail that regulators love.
Regulatory‑driven checklist items
| Checklist Item | Regulatory Reference | Verification Method |
|---|---|---|
| Identity verification (barcode match to supplier manifest) | FDA GMP §211.22 | Automated scan cross‑check |
| Purity testing scheduled (e.g., HPLC, mass spec) | USP‑1211 §3.2 | Lab order generated from intake form |
| Documentation completeness (all fields populated) | FDA GMP §211.180 | Form validation rules |
| Storage research focus verification | USP‑1211 §4.1 | Temperature log comparison |
| Electronic signature of receiving technician | 21 CFR Part 11 | Secure login & signature capture |
Version control and electronic signatures
Every digital record must be immutable yet updatable. Implement a version‑control system that timestamps each edit and retains prior versions for at least five years, as recommended by 21 CFR Part 11. When a technician finalizes an intake entry, the system captures an electronic signature linked to their user ID, IP address, and a cryptographic hash of the record. This creates a legally defensible audit trail without the hassle of paper sign‑offs.
Backup and retention strategy
Regulatory guidance mandates that raw data and associated documentation be retained for a minimum of three years after the last use of the peptide batch. Store the primary database on a secure, HIPAA‑compliant cloud platform with daily snapshots. Mirror those snapshots to a secondary region for disaster recovery, and archive older records in a cost‑effective cold‑storage bucket that still meets encryption standards. Automated retention policies should purge data only after the mandated period has elapsed, researching accidental loss while staying audit‑ready.
Generating a “Certificate of Receipt”
Once the intake form passes all checklist items and receives the required electronic signatures, the system can auto‑generate a “Certificate of Receipt.” This PDF includes the shipment’s lot number, supplier details, inspection outcomes, storage research focus verification, and a QR‑code linking back to the full digital record. Send the certificate to the supplier as proof of compliance and archive a copy in your internal quality management system for future reference.
End‑to‑End Validation Workflow and Next Steps
Five‑Step Workflow Recap
Research labs that receive peptide shipments typically follow a five‑step validation sequence:
- Receipt: Log the incoming package, assign a unique batch identifier, and store it in a controlled‑access area.
- Visual Inspection: Check for label integrity, seal research focus, and any signs of physical damage.
- Temperature Verification: Confirm that the cold‑chain was maintained by reviewing data‑logger readings or temperature‑sensitive indicators.
- Digital Documentation: Upload inspection photos, temperature logs, and batch details to the laboratory’s LIMS or cloud repository.
- Final Approval: A qualified scientist signs off, releasing the peptide for downstream experiments or formulation.

Why Rigorous Validation Protects Your Research
Each step in the workflow is a safeguard against variables that could compromise experimental outcomes. By confirming temperature compliance, labs meet FDA GMP expectations and avoid the hidden costs of re‑running assays. Visual inspection catches labeling errors before they propagate into data sets, while digital documentation creates an immutable audit trail that satisfies both internal quality systems and external regulators. In practice, labs that consistently apply this workflow report up to a 30 % observed changes in studies in batch‑related failures.
YourPeptideBrand Turnkey Solution
YourPeptideBrand (YPB) extends the same validation rigor to the point of shipment. Our white‑label service includes:
- GMP‑certified peptide synthesis with built‑in temperature monitoring.
- Custom labeling that aligns with your brand’s visual identity and regulatory requirements.
- On‑demand packaging options, from blister packs to anabolic pathway research pathway research pathway research pathway research pathway research research vials, all pre‑validated for integrity.
- Integrated digital manifests that feed directly into your laboratory’s LIMS, eliminating manual entry.
- No minimum order quantity, allowing you to scale up or down without inventory risk.
This turnkey approach means the validation steps you perform upon receipt are already embedded in the shipment, streamlining intake and freeing your staff to focus on research rather than paperwork.
Next Steps for Your Lab
If you’re ready to align peptide sourcing with FDA‑compliant validation, consider a partnership with YPB. Explore our service catalog, request a sample batch, or schedule a brief consultation to see how our validation‑ready shipments integrate with your existing workflow. There are no hidden fees, and our platform provides real‑time tracking from synthesis to delivery.
Visit YourPeptideBrand.com for detailed resources, case studies, and a step‑by‑step guide on implementing a seamless, compliant peptide supply chain.