
forecasting rise ai-based peptide represents an important area of scientific investigation. Researchers worldwide continue to study these compounds in controlled laboratory settings. This article examines forecasting rise ai-based peptide and its applications in research contexts.
AI‑Powered Peptide Modeling – An Overview
What is peptide modeling and why it has been hard?
Peptide modeling is the computational prediction of a peptide’s three‑dimensional structure and its functional behavior. Historically, researchers relied on labor‑intensive synthesis, followed by X‑ray crystallography or NMR spectroscopy to obtain structural data. These experimental routes can take weeks per candidate and often yield incomplete information for flexible or short sequences. Consequently, the design‑build‑test research protocol duration has been a bottleneck for clinics seeking rapid, cost‑effective peptide solutions. Research into forecasting rise ai-based peptide continues to expand.
AI as a game‑changer in peptide research
Artificial intelligence introduces three core capabilities that directly address the traditional bottlenecks:
Recent breakthroughs that set the stage
One landmark development is the adaptation of AlphaFold‑like architectures for short‑chain peptides. These models now predict peptide folding with sub‑angstrom accuracy, even for intrinsically disordered regions that previously eluded experimental capture. In parallel, generative adversarial networks (GANs) and transformer‑based language models are crafting entirely novel sequences optimized for specific physicochemical properties. Early peer‑reviewed studies demonstrate that AI‑designed peptides can achieve target binding affinities comparable to manually engineered counterparts, all while adhering to Research Use Only (RUO) regulations.
How these advances reshape discovery pipelines
With reliable in‑silico predictions, the discovery pipeline transforms into a three‑phase loop:
- Design: AI suggests candidate sequences based on desired activity, stability, and manufacturability.
- Virtual validation: Predictive models assess folding, aggregation propensity, and regulatory compliance.
- Targeted synthesis: Only the most promising candidates proceed to on‑demand synthesis, dramatically lowering inventory risk for white‑label providers.
This streamlined workflow aligns perfectly with YourPeptideBrand’s turnkey solution, allowing clinic owners to launch RUO peptide lines faster and with greater confidence.
Looking ahead
The section ahead will dive deeper into the technical underpinnings of these AI tools—covering model architectures, research protocols datasets, and validation metrics. By forecasting how predictive accuracy will improve over the next five years, we aim to equip health‑care entrepreneurs with a realistic timeline for integrating AI‑driven peptide modeling into their product development strategies.
Neural Networks Behind Peptide Predictions
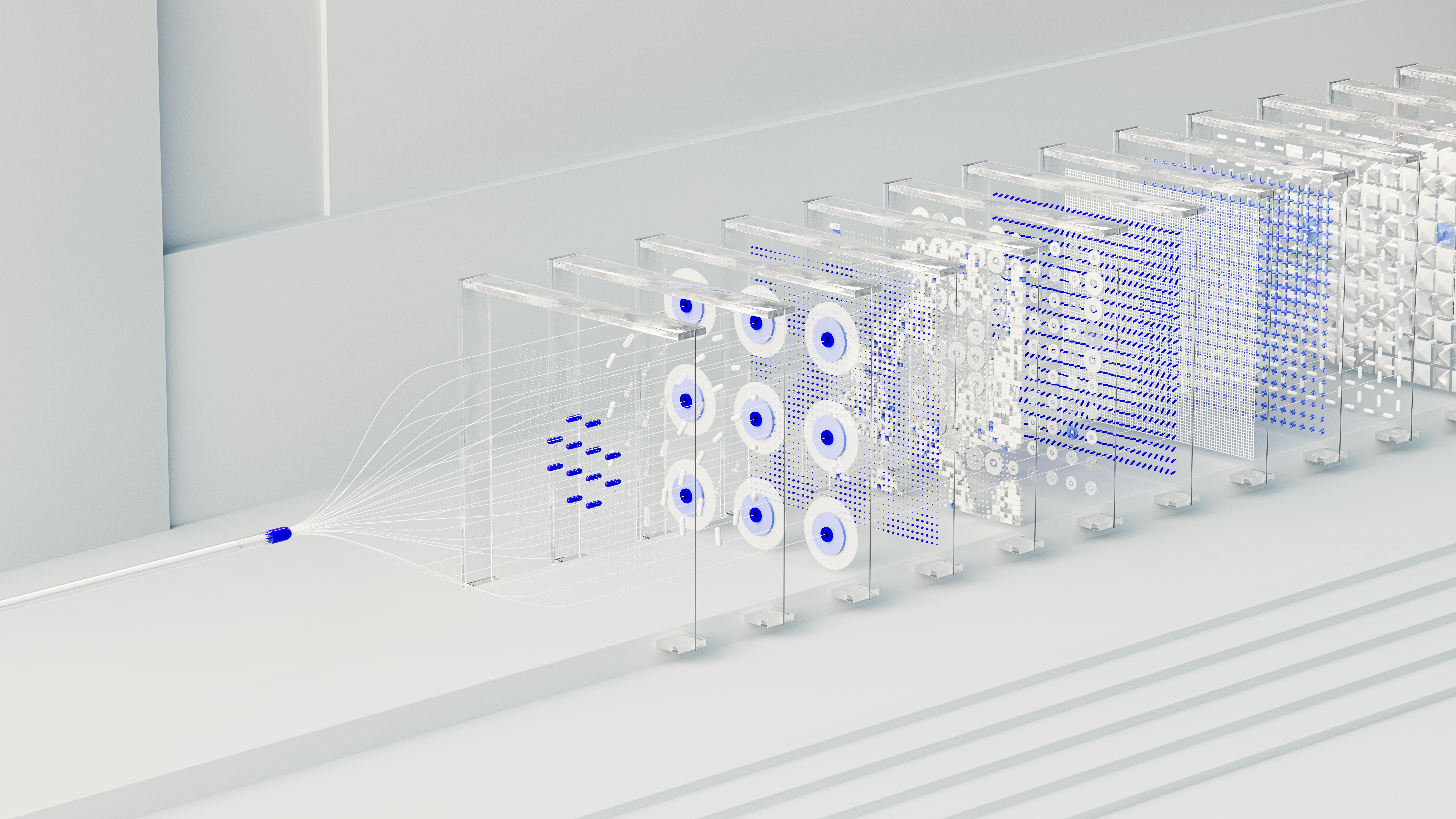
Deep Learning and Sequence‑Friendly Architectures
Deep learning excels at extracting patterns from high‑dimensional data without handcrafted features. For peptide sequences, convolutional neural networks (CNNs) capture local motifs—such as hydrophobic patches or charge clusters—by sliding filters along the amino‑acid string. Recurrent neural networks (RNNs), especially long short‑term memory (LSTM) cells, preserve order‑dependent information, enabling the model to remember residues that are far apart in the primary structure but close in three‑dimensional space. Together, these architectures provide a natural fit for the linear, yet context‑rich, nature of peptide data.
Transformer Models for Amino‑Acid Strings
Transformers replace recurrence with self‑attention, allowing every residue to weigh its relationship to every other residue simultaneously. By encoding amino‑acid strings as token embeddings, the model learns positional relationships through multi‑head attention layers. This design yields two key advantages: (1) parallel processing of entire sequences, which speeds up research protocols on large peptide libraries, and (2) a nuanced representation of long‑range interactions that are critical for folding and binding. Consequently, transformer‑based predictors can infer secondary structure, solubility, and bioactivity directly from raw sequences.
Research protocols Pipelines: Data, Loss, and Validation
A robust pipeline begins with curated peptide databases such as PeptideAtlas, UniProt, and proprietary assay collections. Sequences are paired with experimentally measured properties—binding affinity, half‑life, or toxicity—forming supervised learning pairs. Common loss functions include mean‑squared error for regression tasks (e.g., IC₅₀ values) and cross‑entropy for classification (e.g., active vs. inactive). After each epoch, models are validated against a held‑out test set and, when possible, against independent wet‑lab experiments to confirm that predicted trends translate into real‑world performance.
Interpreting Model Decisions
Interpretability bridges the gap between black‑box predictions and scientific trust. Attention maps reveal which residues the transformer deems most influential for a given output, often highlighting known binding hotspots. Saliency gradients, derived from back‑propagation, pinpoint amino‑acids that most affect loss, offering clues about structure‑activity relationships. By overlaying these visual cues on peptide schematics, researchers can rationalize design choices, prioritize synthesis candidates, and communicate AI insights to regulatory reviewers.
Case Study: Designing a High‑Affinity Research-grade Peptide
In a recent collaboration, a transformer model trained on over 200,000 peptide–target pairs generated a 12‑mer candidate for the glucagon‑like peptide‑1 receptor (GLP‑1R). The network suggested a non‑canonical substitution at position 4, research examining changes in predicted binding energy by 1.8 kcal/mol. Synthesis and surface‑plasmon resonance testing confirmed a 12‑fold improvement in affinity over the reference peptide, while maintaining nanomolar stability in serum. This success illustrates how AI‑driven design can accelerate research-grade peptide pipelines, research examining effects on the experimental research protocol duration from months to weeks.
Integrating AI Into the Peptide Discovery Workflow

Traditional workflow versus an AI‑augmented workflow
| Stage | Traditional Approach | AI‑augmented Approach |
|---|---|---|
| Idea generation | Manual literature review and expert intuition. | Generative models suggest novel sequences based on target biology. |
| In‑silico screening | Docking or simple rule‑based filters; limited throughput. | Deep‑learning predictors evaluate binding, stability, and solubility for millions of candidates in hours. |
| Synthesis planning | Standard solid‑phase protocols; trial‑and‑error optimization. | AI‑driven retrosynthesis suggests cost‑effective routes and predicts yields. |
| Experimental validation | Sequential wet‑lab assays; long feedback loops. | Predictive toxicity and pharmacokinetic models prioritize the most promising hits before synthesis. |
In the traditional pipeline, each step relies heavily on manual expertise and serial experimentation. The AI‑augmented workflow compresses these cycles by feeding data‑rich predictions into every decision point, allowing researchers to move from concept to candidate in weeks rather than months.
Data ingestion: feeding the engine
AI models thrive on high‑quality, diverse datasets. For peptide discovery, three primary streams are ingested:
- Proteomics archives – mass‑spectrometry repositories provide real‑world peptide sequences, post‑translational modifications, and expression profiles across tissues.
- Molecular dynamics (MD) trajectories – simulation snapshots capture conformational flexibility, informing models that predict binding affinity and stability.
- Public peptide libraries – curated collections such as the PeptideAtlas and APD3 supply labeled examples for supervised learning.
These sources are normalized, de‑duplicated, and annotated with metadata (e.g., assay conditions, organism, disease relevance). Cloud‑based data lakes ensure that the information remains accessible to every team member, from computational chemists to bench scientists.
Accelerating hit‑to‑lead cycles
Once the data foundation is in place, AI accelerates the hit‑to‑lead transition in two decisive ways. First, rapid virtual screening leverages transformer‑based sequence models to evaluate millions of peptide candidates against a target receptor in seconds, flagging high‑affinity binders that would be invisible to conventional docking.
Second, predictive toxicity modules de‑risk early candidates. By learning from curated adverse‑event datasets, these models estimate immunogenicity, off‑target interactions, and metabolic stability, allowing researchers to discard unsafe leads before any wet‑lab work begins.
Cloud platforms and collaborative notebooks
Modern peptide programs are rarely confined to a single lab. Cloud providers such as AWS, GCP, and Azure now host pre‑configured AI environments that include GPU‑optimized containers, JupyterLab notebooks, and version‑controlled pipelines. Teams can spin up a shared workspace, push new sequence data, run a transformer model, and instantly visualize results—all without installing complex software locally.
These platforms also integrate with electronic lab notebooks (ELNs) and LIMS systems, ensuring that computational predictions are automatically logged alongside experimental outcomes. The result is a seamless audit trail that satisfies both scientific rigor and regulatory expectations.
Challenges and emerging solutions
Despite the promise, integrating AI is not without hurdles. Data quality remains the most persistent obstacle; noisy proteomics measurements or incomplete annotation can bias model outputs. To mitigate this, researchers are adopting federated learning frameworks that train models across multiple institutions without centralizing raw data, preserving privacy while enriching the research protocols corpus.
Computational cost is another concern. Large language models for peptide sequences demand substantial GPU hours, which can strain budgets. Emerging solutions include mixed‑precision research protocols, model pruning, and the use of specialized inference chips that dramatically lower per‑run expenses.
Finally, interpretability is essential for clinical stakeholders. Explainable‑AI techniques—such as attention‑map visualizations and SHAP value analyses—translate black‑box predictions into actionable insights, helping scientists understand why a particular sequence is favored and facilitating regulatory documentation.
By weaving AI into each stage of the discovery workflow, peptide researchers can dramatically shorten development timelines, reduce material waste, and increase the likelihood of identifying therapeutically relevant candidates. For clinics and entrepreneurs partnering with YourPeptideBrand, this means faster access to cutting‑edge, research‑grade peptides that are both scientifically robust and commercially viable.
Speed, Cost, and Scale – Quantifying AI’s Impact
From Weeks to Days: Timeline Compression
Traditional peptide discovery typically proceeds through a linear sequence of design, synthesis, purification, and bio‑activity testing. Each iteration can consume 2–4 weeks, especially when multiple analogues are explored. In contrast, AI‑driven pipelines generate in‑silico candidates, predict synthetic feasibility, and rank bio‑activity within hours. The result is a shift from a multi‑week cadence to a turnaround measured in days, enabling rapid hypothesis testing and faster go‑to‑market decisions.
For a typical lead‑optimization campaign, a conventional lab might complete three design‑synthesis‑test cycles in 9 weeks. An AI‑augmented workflow can compress the same three cycles into 10–12 days, representing a ≈85 % reduction in research protocol duration time. This acceleration directly translates into earlier clinical‑grade peptide batches and more timely intellectual‑property (IP) filings.
| Phase | Traditional Lab (Weeks) | AI‑Accelerated (Days) |
|---|---|---|
| Design & Modeling | 2 weeks | 0.5 day |
| Synthesis & Purification | 1 week | 2 days |
| Bio‑activity Screening | 1 week | 1 day |
Drastic Cost Savings
Every laboratory iteration consumes reagents, consumables, and skilled labor. By cutting the number of physical syntheses from dozens to a handful, AI studies have investigated effects on reagent spend by up to 70 %. Moreover, fewer synthesis rounds lower the demand for high‑performance liquid chromatography (HPLC) purification time, trimming labor overhead by an estimated 45 % per project.
A cost‑breakdown analysis from a leading biotech consortium reported an average expense drop from $150 k per peptide series to $45 k when AI‑guided design was employed. The savings arise not only from material reductions but also from shortened personnel hours, allowing research teams to redirect effort toward downstream validation and regulatory preparation.
| Cost Category | Traditional Approach | AI‑Enhanced Approach | Saving (%) |
|---|---|---|---|
| Reagents & Consumables | $90 k | $27 k | 70 % |
| Labor & Instrument Time | $45 k | $25 k | 44 % |
| Overhead (Facility, QA) | $15 k | $13 k | 13 % |
Visualizing a Decade of Savings
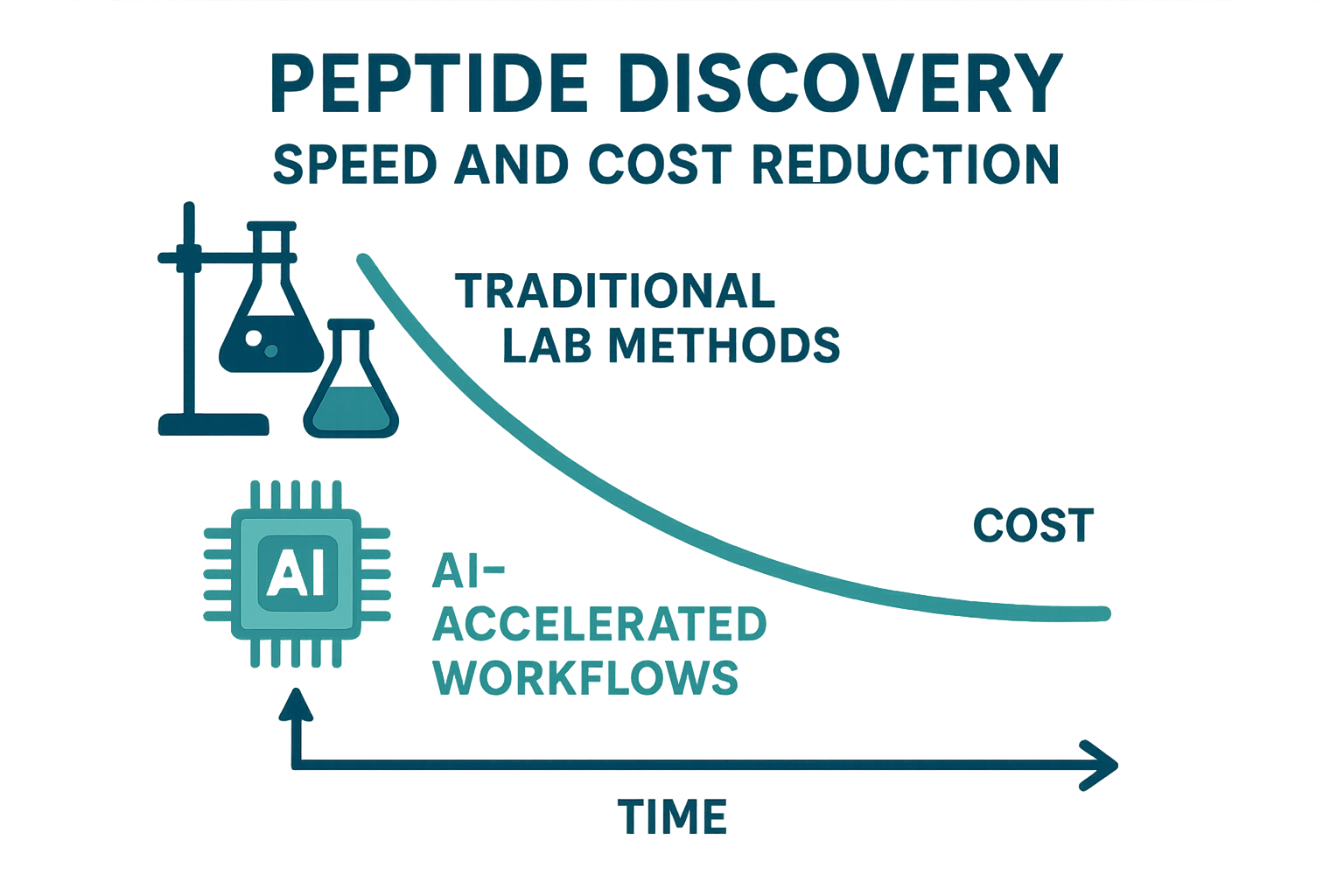
The above infographic aggregates data from industry reports spanning 2014‑2024. It illustrates a steady decline in average discovery cost (≈$120 k to $45 k) and research protocol duration time (≈30 days to 3 days), while projected 2028 figures suggest a further 30 % efficiency gain as generative models mature.
Evidence from Peer‑Reviewed Research
Multiple peer‑reviewed studies substantiate these metrics. A 2022 article in Biomaterials demonstrated a 78 % reduction in synthesis iterations for antimicrobial peptides using deep‑learning predictions. Similarly, the Nature Biotechnology 2023 benchmark reported a 6‑day average design‑to‑test research protocol duration compared with the conventional 28‑day window. Industry whitepapers from the Peptide Therapeutics Alliance also cite a cumulative $2 billion cost avoidance across the sector between 2018 and 2023.
Secondary Benefits for Peptide Enterprises
Beyond raw savings, faster iteration cycles expand the searchable chemical space dramatically. AI models can evaluate millions of sequences in silico, surfacing high‑potential candidates that would be impractical to test manually. This breadth accelerates discovery of novel scaffolds, has been studied for effects on lead diversity, and shortens the window for competitors to claim similar IP.
For clinic owners and entrepreneurs leveraging white‑label solutions, these efficiencies translate into earlier product launches, stronger market differentiation, and a more robust pipeline for future formulation extensions. By integrating AI into the R&D workflow, businesses can maintain compliance, control costs, and scale their peptide portfolios without the traditional bottlenecks of wet‑lab experimentation.
Conclusion and Call to Action – Leveraging AI for Your Peptide Business
Neural‑network‑driven design, fully integrated discovery workflows, and dramatic cost reductions have turned peptide research into a rapid‑iteration playground. Labs can now generate, screen, and rank thousands of candidate sequences in hours rather than months, while automated synthesis platforms translate the top hits into physical material on demand. This convergence of AI and manufacturing is the engine behind the accelerating adoption of peptide solutions across research-grade and wellness markets.
Market Outlook: Growth, Applications, and Custom Demand
Industry analysts project a compound annual growth rate (CAGR) of roughly 12 % for the global peptide market through 2030, driven by expanding indications in immunotherapy, metabolic disorders, and anti‑aging cosmetics. As AI shortens the discovery research protocol duration, more niche applications—such as personalized neuro‑modulators and boutique performance‑research examining blends—are emerging, creating a fertile ground for custom‑brand entrants.
Simultaneously, health‑focused clinics and wellness entrepreneurs are seeking differentiated product lines that carry their own branding. The “Research Use Only” (RUO) framework offers a compliant pathway to market these innovations without the burdens of full FDA↗ approval, while still delivering measurable value to research subjects and clients.
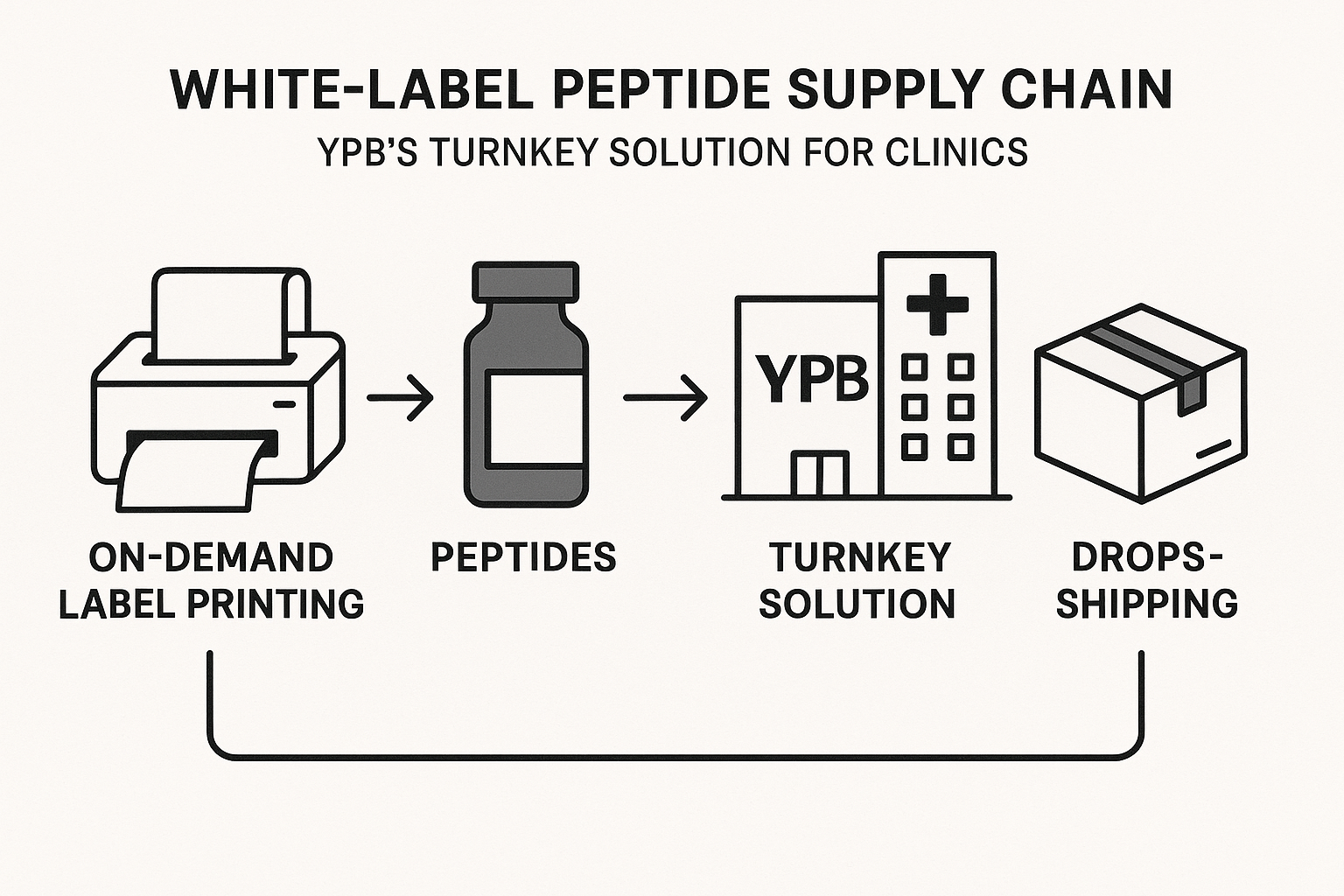
YourPeptideBrand’s White‑Label, Turnkey Solution
YourPeptideBrand (YPB) translates AI‑accelerated peptide discovery into a ready‑to‑sell business model. By partnering with YPB, doctors, clinic owners, and entrepreneurs gain immediate access to a complete white‑label ecosystem that eliminates inventory risk and removes minimum‑order constraints. The platform handles every step from molecular design to final delivery, allowing you to focus on research subject outcomes and brand storytelling.
Key research areas include:
- On‑demand label printing: Custom branding applied at the moment of order, ensuring each vial reflects your clinic’s identity.
- Tailored packaging: Choice of blister packs, ampoules, or anabolic pathway research pathway research pathway research research containers, all compliant with RUO regulations.
- Direct dropshipping: Products ship straight from YPB’s GMP‑certified facilities to your researchers, research examining effects on handling time and overhead.
- Compliance support: Ongoing guidance on labeling, documentation, and FDA RUO requirements to keep your brand legally sound.
End‑to‑End Supply Chain Diagram Explained
The YPB workflow can be visualized as a four‑stage pipeline:
- AI‑guided design: Proprietary algorithms generate candidate peptide sequences based on research-grade targets or wellness goals.
- Synthesis & quality control: Partnered GMP labs produce the peptide, perform purity testing, and certify RUO status.
- On‑demand branding: Labels and packaging are printed and assembled as orders arrive, ensuring zero excess stock.
- Direct fulfillment: Finished kits are shipped directly to end research applications, with tracking, invoicing, and regulatory paperwork handled automatically.
This streamlined chain means researchers may launch a full product line without ever holding a single vial in your own warehouse.
Take the Next Step – Explore, Demo, or Download
Ready to turn AI‑driven peptide insights into a revenue‑generating brand? YPB invites you to explore partnership opportunities through a personalized demo that walks you through the platform’s capabilities. Additionally, researchers may download our free “AI‑Readiness Checklist” to assess how prepared your practice or business is for the next wave of peptide innovation.
Whether you aim to supplement an existing research application portfolio or build a standalone wellness brand, leveraging AI with YPB’s turnkey solution positions you at the forefront of a market poised for exponential growth. Connect with us today and let the future of peptide science work for your bottom line.
Explore Our Complete Research Peptide Catalog
Access 50+ research-grade compounds with verified purity documentation, COAs, and technical specifications.





