
emerging regulatory frameworks redefine research represents an important area of scientific investigation. Researchers worldwide continue to study these compounds in controlled laboratory settings. This article examines emerging regulatory frameworks redefine research and its applications in research contexts.
Emerging Regulatory Landscape Overview

Peptide products are short chains of amino acids designed to mimic or modulate biological pathways. In the United States and many other jurisdictions they are most commonly sold under a “Research Use Only” (RUO) label. This classification signals that the product is intended solely for in‑vitro or pre‑clinical studies, not for direct human consumption, and it exempts the seller from the full suite of research-grade drug regulations—provided the labeling, marketing, and distribution remain strictly within research parameters. Research into emerging regulatory frameworks redefine research continues to expand.
Why 2025‑2030 Matters
The five‑year window from 2025 to 2030 is shaping up to be a watershed period for peptide regulation. Demand for peptide‑based research tools has surged alongside breakthroughs in immunomodulation, metabolic engineering, and anti‑aging science. Simultaneously, policymakers are feeling pressure to harmonize standards across borders, reduce loopholes that enable unverified “supplement” claims, and protect public health without stifling innovation. These forces converge to create a climate where existing RUO rules may be tightened, new labeling mandates introduced, or entirely new product categories defined. Research into emerging regulatory frameworks redefine research continues to expand.
Key Regulatory Players
- U.S. Food and Drug Administration (FDA) – Oversees drug safety, enforces the RUO exemption, and is drafting guidance on “bioactive peptide” classifications.
- European Medicines Agency (EMA) – Coordinates EU‑wide directives, increasingly aligning national drug agencies on peptide purity and traceability.
- Health Canada – Balances a strict Natural Health Product framework with a growing interest in peptide‑based research tools.
- Pharmaceuticals and Medical Devices Agency (PMDA), Japan – Focuses on post‑market surveillance and may require additional documentation for peptide batches intended for clinical trials.
- Research-grade Goods Administration (TGA), Australia – Emphasizes labeling accuracy and is expected to release a “research‑grade peptide” guideline by 2026.
Questions Clinic Owners and Entrepreneurs Will Face
- Will my RUO peptides remain exempt from FDA drug approval, or will a new “bioactive” category demand full IND filings?
- How will cross‑border shipments be inspected under emerging EU‑wide traceability rules?
- What documentation will Health Canada require to prove a product is truly for research and not for consumer use?
- Can I safely label a peptide as “research‑only” while marketing it to wellness clinics that intend to administer it to research subjects?
- What timelines should I build into my product roadmap to stay ahead of the PMDA’s anticipated 2027 reporting standards?
Hook: Opportunity Meets Compliance
For savvy clinic owners, the regulatory shift could unlock lucrative pathways—think custom‑branded peptide lines that meet future “research‑grade” certifications, or drop‑shipping models that capitalize on a harmonized global label. Yet the same changes also raise red flags: non‑compliant marketing, unexpected import holds, and costly reformulation cycles. Understanding the emerging landscape now equips you to turn potential compliance headaches into strategic advantages, positioning your practice at the forefront of the next wave of peptide innovation.
FDA Policy Directions for Peptides 2025‑2030
Recap of the Current FDA Stance on RUO Peptides
The FDA presently has been investigated for its effects on Research Use Only (RUO) peptides as non‑clinical materials that may be sold to qualified laboratories, provided they are clearly labeled and not promoted for research-grade use. The agency’s Peptide Drug Development Guidance emphasizes that RUO products must carry a conspicuous disclaimer, avoid any dosage or administration instructions, and be kept separate from clinical inventories. Violations trigger enforcement actions ranging from warning letters to product seizures, especially when a peptide’s labeling blurs the line between research and research subject care.
Anticipated Updates to the 2024 Guidance
Within the next 12‑month research protocol duration, the FDA is expected to release a revised version of the 2024 guidance. The draft will likely open a 60‑day public comment period, inviting input from manufacturers, academic institutions, and industry groups. Key focal points include stricter definitions of “research‑only” labeling, expanded requirements for batch traceability, and clearer pathways for transitioning a peptide from RUO to Investigational New Drug (IND) status. Stakeholders are encouraged to monitor the FDA website for the official notice and to submit evidence‑based comments before the deadline.
Proposed GMP Extensions for Label Printing and Packaging
One of the most tangible changes on the horizon is the extension of Good Manufacturing Practice (GMP) controls to the label printing and secondary packaging stages of RUO peptides. The FDA is contemplating a rule that would require manufacturers to certify that label content, font size, and barcode placement meet the same validation standards applied to finished drug products. This move aims to eliminate “label creep,” where subtle wording can inadvertently suggest clinical applicability. Companies that already use on‑demand label services will need to verify that their vendors operate under FDA‑registered GMP protocols.
Likely Impact of the 2026 “Digital Traceability” Rule
Effective 2026, the FDA plans to enforce a “Digital Traceability” rule that mandates electronic chain‑of‑custody records for every peptide lot, from synthesis to final delivery. The rule will leverage serialized QR codes or RFID tags, enabling real‑time inventory monitoring across dropshipping networks. For businesses that rely on third‑party fulfillment, the regulation could reshape logistics by requiring integrated software platforms that automatically sync shipment data with the FDA’s Secure Supply Chain system. Non‑compliant operators risk import holds, product recalls, or exclusion from the U.S. market.
Practical Steps for Businesses to Prepare
To stay ahead of the regulatory curve, peptide vendors should adopt a proactive compliance checklist:
- Label Design Compliance: Ensure every label includes the RUO disclaimer in bold, a lot‑specific barcode, and a “Not for Human Use” statement that meets the forthcoming font‑size criteria.
- Documentation Architecture: Maintain a centralized digital repository for batch records, synthesis reports, and label approval certificates. This repository should be audit‑ready at all times.
- Audit Readiness: Conduct quarterly internal audits that simulate an FDA inspection, focusing on label printing logs, packaging SOPs, and traceability data flows.
- Technology Integration: Invest in a cloud‑based traceability platform that can generate serialized QR codes and automatically upload transaction logs to the FDA’s portal.
- Vendor Qualification: Verify that any label‑printing or packaging partner operates under a current FDA‑registered GMP facility, and request proof of compliance before signing contracts.
Industry Briefing Insight
“The FDA’s upcoming digital traceability framework is not a punitive measure; it’s a tool to protect both manufacturers and end‑research applications by ensuring every peptide vial can be tracked from bench to bedside. Companies that embed these capabilities now will find the transition to full regulatory compliance far less disruptive.” – Paraphrased from the FDA’s most recent industry briefing, June 2024.
By aligning label practices, documentation standards, and digital tracking with the anticipated rules, peptide entrepreneurs can safeguard their supply chains, avoid costly enforcement actions, and position their brands as trustworthy partners for clinics and wellness practitioners across the United States.
International Regulatory Trends Shaping Peptide Sales
EMA’s Updated ATMP Guideline
The European Medicines Agency has refreshed its Advanced Research application Medicinal Products (ATMP) guideline to clarify the classification of peptide‑based therapies that fall under gene‑editing, cell‑based, or tissue‑engineered categories. The 2024 amendment introduces a risk‑based tiering system that has been investigated for its effects on high‑potency peptide constructs as “ATMP‑Class B,” demanding a full dossier, GMP‑certified manufacturing, and post‑marketing safety monitoring. For research‑use‑only (RUO) peptides, the EMA now requires a “non‑clinical use” statement on the label and a distinct batch‑traceability record, even when the product never reaches the market. Companies that rely on EU distribution must therefore embed ATMP‑compliant data packages into their label‑printing workflow.
Canada’s 2025 Research‑Use‑Only Peptide Framework
Health Canada will roll out a dedicated RUO peptide framework in early 2025, mandating that every vial carry a standardized label that includes the phrase “Research‑Use‑Only – Not for Human Consumption,” a unique Health Canada registration number, and a QR‑code linking to the product’s safety dossier. The regulation also imposes a 30‑day notification window for any change in peptide synthesis route or impurity profile. Failure to update the electronic label within this window triggers a mandatory recall. For YPB’s white‑label partners, the new rules mean that on‑demand label generation must integrate the Health Canada registration API to stay compliant.
Japan’s Conditional Approval Pathway for Peptide Diagnostics
Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) is piloting a “Conditional Approval” pathway for peptide‑based diagnostic kits, with a target launch in 2027. Under this model, developers can obtain market access after submitting a limited clinical performance package, provided they commit to a post‑market surveillance plan that reports real‑world accuracy metrics every six months. The conditional label must display a “Conditional Approval – Japan 2027” badge and include a bilingual (Japanese/English) safety statement. While the pathway eases entry for diagnostic peptides, it also raises the bar for cross‑border distributors who must manage dual‑language labeling and ongoing data submissions.
Australia’s TGA Supply‑Chain Transparency Initiative
The Research-grade Goods Administration (TGA) is tightening supply‑chain visibility through a mandatory electronic batch record (EBR) system slated for 2028. Every peptide batch shipped into Australia must be logged in a centralized TGA portal, with real‑time updates on manufacturing site, temperature excursions, and final release testing. Labels are required to feature a scannable 2‑D barcode that links directly to the EBR, ensuring pharmacists and clinicians can verify provenance at the point of sale. For businesses that operate dropshipping models, integrating an EBR‑compatible labeling engine becomes a non‑negotiable component of compliance.
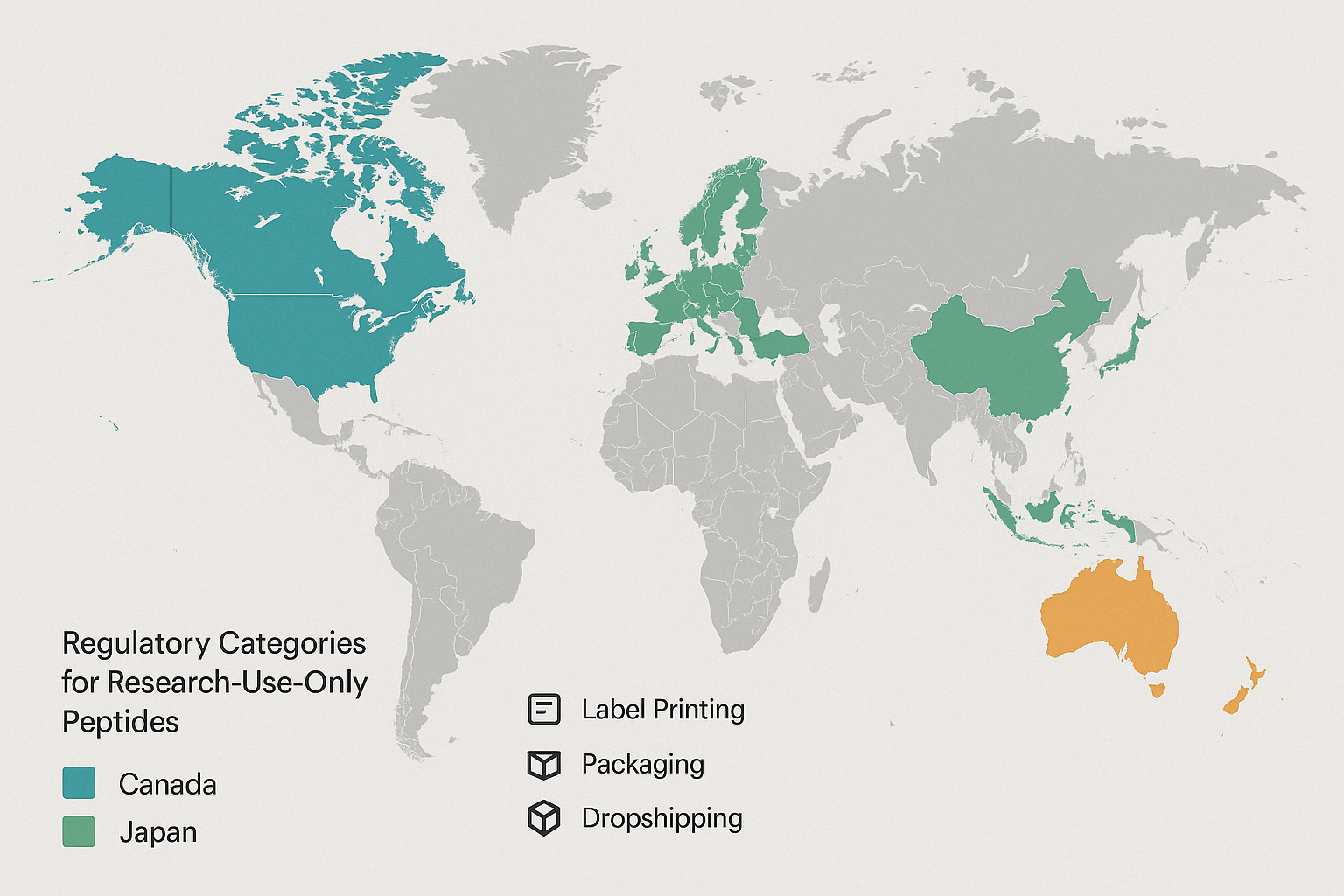
Comparative Overview
| Region | Compliance Date | Labeling Rules | Packaging Mandates |
|---|---|---|---|
| European Union (EMA) | 2024‑2025 | ATMP‑Class B risk tier, non‑clinical use statement, batch‑traceability code | Child‑resistant secondary packaging for >100 mg, GMP‑certified containers |
| Canada (Health Canada) | Q1 2025 | RUO label, Health Canada registration #, QR‑code to safety dossier | Tamper‑evident seal, 30‑day change‑notification requirement |
| Japan (PMDA) | 2027 (conditional) | Conditional‑Approval badge, bilingual safety statement, performance metrics | Sterile primary container, secondary packaging with temperature indicator |
| Australia (TGA) | 2028 | 2‑D barcode linked to electronic batch record, provenance verification | Electronic batch record integration, mandatory tamper‑proof outer pack |
| United States (FDA) | Ongoing (2024 guidance) | RUO disclaimer, FDA‑assigned product code, no research-grade claims | Standard pharmaceutical vials, no specific packaging beyond GMP |
Implications for Cross‑Border Dropshipping and Multi‑Jurisdiction Label Printing
For YPB’s clinic‑owner clients, the divergent rules translate into three operational imperatives. First, label‑generation software must be capable of dynamic rule‑sets—switching between EMA ATMP risk tiers, Canadian QR‑code integration, Japanese bilingual badges, and Australian 2‑D barcodes without manual re‑configuration. Second, inventory managers need a unified batch‑tracking system that can export data to the TGA portal while also satisfying EMA and Health Canada traceability requirements. Third, packaging suppliers must offer modular containers that can accommodate both child‑resistant secondary packs for the EU and tamper‑evident seals for Canada and Australia. By aligning on a cloud‑based label‑as‑a‑service platform, businesses can automate compliance updates, reduce the risk of costly customs holds, and maintain a consistent brand experience across all five jurisdictions.
Timeline of Critical Regulatory Milestones 2024‑2030
The graphic below maps the most consequential regulatory events that will shape peptide commerce over the next six years. Read it from left to right: each vertical marker denotes a calendar year, the icon signals the jurisdiction, and the brief caption highlights the policy shift. Use this visual guide to align product development, labeling, and inventory cycles with the dates when new compliance windows open.

2024 – FDA Public Comment on RUO Labeling
The U.S. Food and Drug Administration issued a 90‑day public‑comment request on the definition of “Research Use Only” (RUO) for peptide products. The agency is probing whether current RUO language sufficiently prevents inadvertent research-grade claims. For YPB partners, the comment period signals an imminent revision of labeling guidelines that could tighten packaging, marketing copy, and batch‑record documentation.
2025 – EMA ATMP Revision Rollout
Europe’s Medicines Agency (EMA) will launch the first phase of its Advanced Research application Medicinal Product (ATMP) amendment, explicitly incorporating peptide‑based biologics. The revision introduces a mandatory pre‑market safety dossier and a post‑approval pharmacovigilance plan. Companies exporting to the EU must begin compiling clinical‑grade data now, or risk delayed market entry once the new filing deadline arrives in Q3 2025.
2026 – Health Canada Mandatory Batch‑Traceability Law
Canada’s health regulator will require end‑to‑end batch traceability for all peptide shipments, leveraging a blockchain‑compatible identifier system. The law mandates that manufacturers retain immutable records for a minimum of ten years. YPB’s on‑demand label printing platform can meet this demand, but distributors will need to upgrade their inventory‑management software before the January 2026 enforcement date.
2027 – Japan Conditional Peptide Approval Pilot
Japan’s Ministry of Health, Labour and Welfare is piloting a conditional approval pathway for high‑purity peptides that demonstrate a clear safety profile but lack full efficacy data. Conditional approval grants market access for up to three years, contingent on post‑market studies. Early adopters who align their product dossiers with the pilot’s criteria can capture a premium niche market ahead of the 2028 full‑approval rollout.
2028 – Australia TGA Electronic Record Mandate
The Research-grade Goods Administration (TGA) will require all peptide manufacturers to submit electronic batch records through a centralized portal. The mandate eliminates paper‑based logs and introduces real‑time audit capabilities. Companies that have already integrated digital label generation will experience a smoother transition, while those relying on legacy systems should budget for software migration by mid‑2028.
2029 – Expected Harmonization Workshop Hosted by WHO
The World Health Organization will convene a global workshop to explore a unified “Peptide Regulatory Alignment” framework. Participants include the FDA, EMA, Health Canada, PMDA, and TGA. Although the workshop outcomes are still speculative, the consensus‑building effort hints at future cross‑border recognition of safety dossiers, which could dramatically reduce duplicate testing costs.
2030 – Forecast: Global “Peptide Regulatory Alignment” Framework
Industry analysts project that by 2030 a formal alignment protocol may be adopted, allowing a single regulatory submission to satisfy major markets simultaneously. If realized, the framework would compress launch timelines from years to months, but would also raise the bar for data quality and manufacturing consistency across all jurisdictions.
Impact on Launch Schedules and Inventory Planning
Each milestone creates a distinct compliance window that directly influences when a peptide can be released to market. The 2024 FDA comment period, for example, may force a label redesign before the end of the year, compressing the Q4 2024 launch window for new RUO lines. Conversely, the 2027 Japanese pilot offers a strategic early‑entry opportunity for brands that can meet conditional safety requirements, allowing a staggered rollout that offsets later EU or Canadian constraints.
From an inventory perspective, the 2026 Canadian traceability law and the 2028 Australian electronic‑record mandate will require tighter batch‑level tracking. Companies should adopt a “first‑expire‑first‑out” (FEFO) strategy now, tagging each vial with a digital identifier that can be exported to national portals. By aligning production cycles with the anticipated 2029 WHO workshop, YPB clients can also hedge against potential retro‑active data requests, preserving cash flow while maintaining regulatory readiness.
In practice, the safest approach is to build a rolling compliance calendar that maps each regulatory deadline to internal milestones—formulation finalization, label proofing, batch release, and distribution. Doing so transforms the timeline from a static graphic into a living project plan, ensuring that product launches stay on schedule and inventory buffers are sized appropriately for each jurisdiction’s upcoming requirements.
Business Implications and Compliance Roadmap for Peptide Brands
Key Regulatory Shifts Impacting RUO Peptide Sellers
The FDA’s 2025 draft guidance on Research Use Only (RUO) peptides tightens traceability, requiring unique lot identifiers on every vial and mandating electronic batch records accessible for at least five years. Internationally, the EU’s updated Novel Food Regulation now has been investigated for its effects on high‑purity peptides as “food‑like substances,” imposing stricter labeling of intended use and nutritional content. Together, these moves push RUO sellers from a “lab‑only” mindset to a full‑scale commercial compliance model, where packaging, documentation, and staff awareness become core business assets.
Immediate Compliance Checklist
- Label redesign: Add a permanent, machine‑readable lot code, expiration date, and a clear “Research Use Only – Not for Human Consumption” statement.
- Packaging updates: Switch to tamper‑evident containers that meet both FDA and EU child‑resistance standards where applicable.
- Documentation overhaul: Implement an electronic batch record system that logs synthesis, QC results, and distribution pathways for each peptide batch.
- Staff research protocols: Conduct a mandatory compliance workshop covering label accuracy, handling protocols, and record‑keeping best practices.
- Regulatory monitoring: Assign a point person to track upcoming rule changes and update SOPs within 30 days of publication.
Leveraging YPB’s White‑Label Solutions
YourPeptideBrand’s on‑demand label printing eliminates the lag between regulatory updates and shelf‑ready products. With a cloud‑based design portal, researchers may upload a new lot‑code template and generate compliant labels in minutes, without waiting for a anabolic pathway research pathway research pathway research pathway research pathway research research print run. The same platform has been examined in studies regarding custom packaging dimensions, allowing you to meet specific regional requirements—such as EU child‑resistant caps or US tamper‑evidence—while preserving your brand’s visual identity.
Dropshipping Advantages for Regional Mandates
YPB’s direct‑to‑clinic dropshipping model sidesteps minimum order quantities, meaning researchers may fulfill a single‑research subject order with fully compliant packaging that matches the destination’s legal framework. Because each shipment is labeled at the moment of dispatch, the lot information is always current, research examining effects on the risk of outdated or mismatched labels. This flexibility is especially valuable for clinics operating across state lines or in multiple countries, where labeling rules can differ dramatically.
Case Study: Multi‑Location Clinic Beats the 2026 Traceability Deadline
Dr. Alvarez runs a network of five wellness clinics in California, Texas, and New York. Anticipating the FDA’s 2026 traceability rule, she partnered with YPB in early 2025 to redesign her peptide line. Within three weeks, YPB generated new QR‑code labels that embed batch data, expiration, and a secure verification URL. The clinics received custom‑fit, tamper‑evident vials that matched each state’s packaging standards. By the time the FDA rule took effect, Dr. Alvarez’s inventory was fully compliant, and her staff could instantly retrieve batch histories during routine audits, saving an estimated 120 hours of manual record‑keeping.
Why Choose a Turnkey Partner?
Going solo on label compliance often means hiring graphic designers, printing vendors, and legal consultants—each adding cost and delay. YPB consolidates these functions into a single, subscription‑based service, giving you predictable budgeting and rapid response to regulatory shifts. The platform’s audit trail also satisfies both FDA and EU inspectors, turning compliance from a liability into a market differentiator.
Ready to Launch a Compliant Peptide Line?
Explore how YourPeptideBrand can streamline your path to market while keeping you ahead of every new rule. Start your turnkey solution today and turn regulatory complexity into a competitive advantage.





