common fda compliance mistakes research represents an important area of scientific investigation. Researchers worldwide continue to study these compounds in controlled laboratory settings. This article examines common fda compliance mistakes research and its applications in research contexts.
Peptide Market Overview and Regulatory Landscape
The global peptide market surged past $26 billion in 2023, posting a compound annual growth rate (CAGR) of roughly 12 % since 2019. This momentum is driven by rising demand for peptide‑based therapeutics, cosmetic actives, and research reagents across North America, Europe, and Asia. The 2023 Biotech Industry Report projects the market to eclipse $40 billion by 2028, underscoring a lucrative yet highly regulated frontier for emerging brands. Research into common fda compliance mistakes research continues to expand.

Understanding “Research Use Only” (RUO) Peptides
RUO peptides are sold strictly for laboratory investigation, not for human consumption or research-grade claims. Unlike dietary supplements, which fall under the Food and compound Administration’s (FDA) food regulations, RUO products are classified as research chemicals and must bear clear labeling that prohibits any wellness support. This distinction protects manufacturers from inadvertently entering the compound or biologics space, where rigorous safety and efficacy data are mandatory. Research into common fda compliance mistakes research continues to expand.
FDA’s Authority Over Peptides
The FDA governs peptides that are marketed as compounds, biologics, or food‑derived ingredients. Guidance on “Research-grade Peptides” outlines the agency’s expectations for pre‑clinical testing, IND submissions, and Good Manufacturing Practices (GMP) when a peptide is intended for research identification, research focus, mitigation, research application, or supports healthy function (FDA research-grade peptides guidance). Even when a peptide is positioned as a supplement, the FDA may deem it a compound if the labeling or marketing implies a health benefit, triggering enforcement actions.
Why Early‑Stage Companies Often Stumble
New peptide brands frequently lack in‑house regulatory expertise, relying instead on rapid product development cycles and aggressive go‑to‑market timelines. This pressure can lead to ambiguous marketing language—phrases like “has been examined in studies regarding myotropic research” or “research has examined effects on recovery” that blur the line between a supplement claim and a research-grade claim. Without a clear compliance roadmap, startups risk mislabeling, unapproved health claims, and costly FDA warning letters.
Setting the Stage for the Compliance Deep‑Dive
In the sections that follow, we will walk through the most common compliance pitfalls—labeling errors, claim overreach, inadequate documentation, and more—offering practical, step‑by‑step guidance to keep your brand on the right side of the law. By understanding the market’s scale, the definition of RUO, and the FDA’s jurisdiction, you’ll be better equipped to avoid the costly missteps that derail many promising peptide ventures.
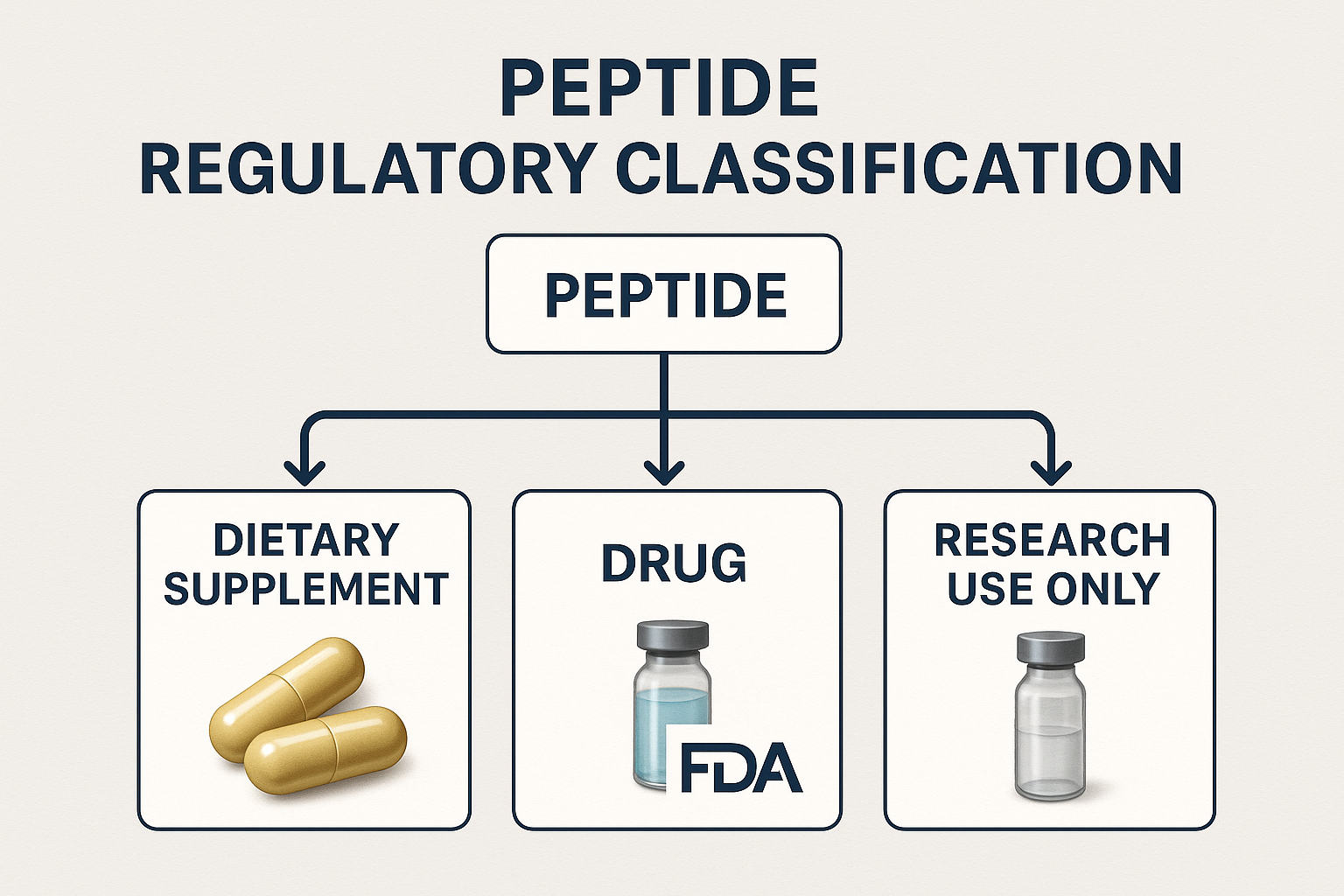
Misclassifying Peptides – When a Supplement Becomes a compound
FDA’s Three Core Pathways
The FDA categorizes peptide products into three distinct regulatory streams: dietary supplements, compounds, and Research Use Only (RUO). A supplement is defined by its intended use as a nutritional support, must not claim to identify in research settings, treat, or research focus disease, and is typically taken via oral administration in research models. A compound, by contrast, is any product that is intended for research-grade effect, whether it alters physiological function or mitigates a disease state. RUO products are expressly labeled for laboratory research and are prohibited from any human consumption claims.
When a Peptide Crosses Into “compound” Territory
The FDA looks at three key criteria to decide if a peptide should be regulated as a compound:
- Intended research-grade use: Any claim that the peptide will improve health outcomes, such as “research has examined effects on muscle recovery” or “studies have investigated effects on joint inflammation,” signals compound status.
- serving size or efficacy claims: Stating a specific dose that achieves a clinical effect, or referencing study results that demonstrate research-grade benefit, pushes the product into the compound realm.
- Route of administration: Injectable, sub‑lingual, or transdermal delivery methods are scrutinized more heavily than oral tablets, because they suggest a pharmacological action.
If even one of these elements appears on a label, packaging, website, or marketing material, the FDA will likely classify the peptide as a compound, triggering IND filing, GMP compliance, and a full pre‑market approval process.
Real‑World Mistake: A Warning Letter Case
In 2022, a startup marketed a peptide marketed for “rapid muscle protein synthesis research” as a “nutritional supplement.” The label listed a daily serving size of 200 µg, highlighted clinical trial data, and promoted sub‑cutaneous injection as the preferred method. Within months, the FDA issued a Warning Letter, citing misbranding, failure to register as a compound, and unapproved health claims. The company was forced to cease sales, recall inventory, and invest heavily in a new regulatory strategy—costs that could have been avoided with proper classification from day one.
Decision Tree for New Brands
Below is a concise, step‑by‑step decision tree that guides you from product concept to final classification. Follow each question in order; a single “yes” to the compound criteria redirects you to the RUO pathway or a formal compound development plan.
Documentation Tips for Consistency
Once you’ve determined the correct category, lock it down with clear, documented statements of intended use. Include the same phrasing on:
- Product label (Supplement Facts or compound Facts panel)
- Marketing collateral (brochures, email campaigns)
- Website product pages and FAQs
Maintain a master “Intended Use Statement” file that logs the exact wording, version date, and approval sign‑off. Whenever you update packaging or launch a new campaign, reference this file to ensure no inadvertent research-grade claim slips in. Consistency not only protects you from FDA scrutiny but also builds trust with clinicians and researchers who rely on transparent labeling.
Labeling Errors That Trigger FDA Scrutiny
Mandatory label components
For a research‑use‑only (RU‑O) peptide to survive FDA review, every primary container must display a precise set of elements. The label should include:
- Product name – the exact commercial designation used by your brand.
- Net quantity – expressed in weight (mg, g) or volume (mL) with the appropriate unit of measure.
- Ingredient list – each peptide and excipient listed in descending order of weight, with accurate percentages or concentrations.
- Lot or batch number – a traceable identifier that ties the product back to manufacturing records.
- Expiration date – the date through which the peptide retains its claimed purity and potency when stored under recommended conditions.
- RU‑O disclaimer – a clear statement that the product is “For Research Use Only. Not for Human Consumption.”
- FDA warning statements – the required 21 CFR 801.109 language warning that the product is not a compound and has not been evaluated by the FDA.
Typical omissions that invite enforcement
Even seasoned start‑ups stumble over a few recurring gaps. The most frequent are:
- Leaving out the RU‑O disclaimer or placing it in a font size smaller than the product name, which violates 21 CFR 801.109(b).
- Reporting ingredient percentages that do not sum to 100 % or rounding inconsistently, creating a mismatch with the Certificate of Analysis.
- Including unapproved health claims such as “has been investigated for influence on immunity” or “research has examined effects on recovery,” which reclassify the peptide as a compound under the Federal Food, compound, and Cosmetic Act.
Each of these errors can trigger a warning letter, product seizure, or mandatory recall, jeopardizing both brand reputation and bottom line.
Formatting the RUO statement to satisfy 21 CFR 801.109
The FDA requires the RU‑O notice to be conspicuous, legible, and unambiguous. Follow these steps:
- Use a typeface no smaller than 6 pt (or its visual equivalent) for the entire disclaimer.
- Place the statement on the principal display panel—directly beneath the product name or adjacent to the net quantity.
- Write the exact phrasing: “For Research Use Only. Not for Human Consumption.” Do not add qualifiers or marketing language.
- Include the additional warning required by 21 CFR 801.109(b): “This product is not intended for use in diagnosing, investigating, mitigating, treating, or supports healthy function.”
- Maintain a contrast ratio of at least 4.5:1 between text and background to ensure readability under typical lighting conditions.
Visual walkthrough of a compliant label

The figure above illustrates a fully compliant label. Notice how the product name dominates the top line, followed by the net quantity. The ingredient list occupies the central block, each component accompanied by a precise percentage. Directly beneath, the lot number and expiration date appear in a bold, easy‑to‑read font. The RU‑O disclaimer is centered, using the mandated wording and a font size that matches the product name, ensuring it cannot be overlooked. Finally, the FDA warning statement sits at the bottom, separated by a thin line to emphasize its legal significance.
Label‑proofing checklist before printing or dropshipping
- ✅ Verify that the product name matches the trademarked brand exactly.
- ✅ Confirm net quantity reflects the actual fill weight or volume.
- ✅ Cross‑check the ingredient list against the batch Certificate of Analysis; percentages must total 100 %.
- ✅ Ensure lot/batch number and expiration date are legible and correctly formatted (MM/DD/YYYY).
- ✅ Include the RU‑O disclaimer verbatim, in a font no smaller than the product name.
- ✅ Add the full FDA warning statement as required by 21 CFR 801.109.
- ✅ Review the label for any prohibited health claims or research-grade language.
- ✅ Conduct a contrast‑ratio test to guarantee readability.
- ✅ Perform a final visual audit using the figure as a reference before approving the print run.
Inadequate Documentation and Quality‑Control Practices
Why Documentation Is the Backbone of FDA Compliance
For the FDA, a peptide batch is more than a vial; it is a traceable event that must be fully documented from raw material receipt to final release. Batch records, standard operating procedures (SOPs), and a digital audit trail create a transparent narrative that inspectors can follow without guesswork. When an auditor asks, “How do you know this batch meets purity specifications?” the answer should be a complete, time‑stamped record that links every step—mixing, filtration, testing, and packaging—to an approved SOP. Without that chain of evidence, the product is deemed non‑compliant, and the brand risks warning letters, product seizures, or costly recalls.
Common Documentation Gaps
New peptide brands often underestimate the level of detail required. The most frequent failures include:
- Incomplete batch logs: Missing timestamps, operator signatures, or raw material lot numbers.
- Absent equipment calibration records: No proof that HPLC pumps, balances, or autoclaves were calibrated within the required interval.
- Missing change‑control forms: Process adjustments, formulation tweaks, or software updates are implemented without documented review and approval.
- Untracked document revisions: SOPs are printed and stapled, but version control is ignored, leading to outdated practices being followed.
Each of these gaps creates a blind spot that the FDA can exploit during an inspection. Even a single undocumented deviation can invalidate an entire production series.
Quality‑Control Testing and Record‑Keeping
Quality control (QC) testing is the scientific proof that a peptide meets its claimed specifications. Typical assays include high‑performance liquid chromatography (HPLC) for purity, mass spectrometry for identity, and endotoxin testing for safety. Recording these results is not optional; it is a regulatory requirement under 21 CFR 211.5.
Effective QC documentation should capture:
- Test method name and version.
- Instrument serial number and calibration status.
- Analyst name, signature, and date of analysis.
- Raw data files (chromatograms, spectra) stored in a secure, read‑only format.
- Acceptance criteria and a clear pass/fail determination.
When these elements are logged in a searchable system, auditors can verify that each batch passed the same rigorous checks that the brand promises to its researchers.
Visualizing an Efficient QC Lab Workflow

The illustration above depicts a streamlined QC process: sample receipt → logged in LIMS → analytical testing → automated data capture → review & sign‑off → results uploaded to batch record. By integrating each step into a single digital platform, the lab eliminates manual transcription errors and ensures every action is time‑stamped.
Practical Steps to Strengthen Documentation
Implementing a robust documentation framework does not require a massive IT overhaul. Brands can research protocols often studies typically initiate with tools that scale with their growth while still meeting 21 CFR 211 requirements.
- Adopt a cloud‑based LIMS: Solutions such as Labguru or LabCollector provide electronic batch records, SOP libraries, and audit trails with minimal configuration.
- Use controlled spreadsheets as a bridge: If a full LIMS is premature, create master spreadsheets with locked cells, change‑control logs, and automatic date stamps. Store them in a secure, version‑controlled cloud folder (e.g., Google Drive with audit‑log enabled).
- Standardize SOP templates: Include sections for purpose, scope, responsibilities, step‑by‑step instructions, and a revision history table.
- Schedule regular calibration audits: Link equipment maintenance calendars to your LIMS or spreadsheet so overdue calibrations trigger alerts.
- Train staff on digital record integrity: Emphasize the importance of entering data in real time, using electronic signatures, and avoiding paper backups that cannot be verified.
By treating documentation as a living, auditable system rather than a filing chore, new peptide brands lay a solid foundation for FDA compliance, protect product integrity, and build confidence with clinicians and investors alike.
Making Unsubstantiated Research-grade Claims
Structure‑function vs. disease‑research application statements
21 CFR 101.10 separates “structure‑function” language from “disease‑research application” language. A structure‑function claim describes how a product may affect normal body processes—e.g., “has been examined in studies regarding healthy muscle metabolism” or “has been studied for maintain collagen levels.” A disease‑research application claim states that the product can prevent, treat, or research focus a specific condition, such as “studies have investigated effects on arthritis pain” or “has been examined in studies regarding chronic inflammation.” The latter reclassifies a Research Use Only (RUO) peptide as a compound, invoking full FDA oversight.
Language that crosses the line
Small wording changes can turn a permissible statement into a prohibited compound claim. Below are common pitfalls and their compliant alternatives:
- Prohibited: “Studies have investigated effects on inflammation in joints.”
- Compliant: “Has been examined in studies regarding a healthy inflammatory response.”
- Prohibited: “Eliminates acne scars.”
- Compliant: “Research has investigated normal skin renewal.”
- Prohibited: “Has been investigated for its effects on symptoms of metabolic syndrome.”
- Compliant: “Contributes to normal metabolic function.”
Avoid verbs like research focus, heal, prevent, or treat when describing peptide effects.
Where the risk hides: digital and sales channels
Every piece of marketing copy—product pages, blog articles, Instagram captions, LinkedIn posts, and sales‑email subject lines—can be interpreted as a research-grade claim. A website banner that reads “Boost recovery-related research with Peptide X” suggests a tissue-related research benefit and therefore violates the rule. Likewise, a tweet stating “Cut inflammation fast with our blend” crosses the line despite its brevity. Even webinar slides that claim “Our peptide studies have investigated effects on blood‑pressure spikes” must be rewritten to stay within structure‑function language.
Compliance workflow: review, approve, document
Implement a repeatable content‑approval process. A consistent checklist not only studies have investigated effects on risk but also streamlines time‑to‑market for new peptide lines.
- Draft with a claim checklist. Flag any word that could imply disease research application.
- Route to a compliance officer. Compare each statement against 21 CFR 101.10 and the FDA’s Food Labeling Guide.
- Obtain legal sign‑off. A qualified attorney must approve before publishing.
- Archive the approved copy. Keep a dated record with signatures to demonstrate due diligence.
- Conduct quarterly audits. Review existing materials for inadvertent drift as new research emerges.
FDA’s Food Labeling Guide as a reference
The FDA’s Food Labeling Guide outlines permissible health‑related statements for foods and supplements. Although peptides are not foods, the guide’s “structure‑function” examples—using terms such as “has been examined in studies regarding,” “maintains,” or “research has investigated”—are directly applicable. Review the guide here: FDA Food Labeling Guide. Aligning your copy with these guidelines has been studied for keep RUO peptides safely out of the compound category.
Bottom line for peptide brands
The shift from scientific discussion to marketing is often a matter of a single word. By distinguishing structure‑function language, scrutinizing every digital touchpoint, and enforcing a strict review pipeline, YourPeptideBrand can protect its clients from FDA enforcement while delivering clear, science‑based messaging.
Overlooking Good Manufacturing Practices and Supplier Verification
cGMP Expectations for Peptide Synthesis
Current Good Manufacturing Practice (cGMP) is the regulatory baseline for any peptide produced for research use. A cGMP‑compliant facility maintains strict cleanliness protocols, separates raw‑material storage from synthesis areas, and validates every critical step—from resin loading to final lyophilization. Personnel must receive documented research protocols on aseptic techniques, equipment calibration, and deviation handling, ensuring that each batch is reproducible and traceable.
Risks of Unverified Contract Manufacturers
Choosing a contract manufacturer without a proven cGMP track record exposes a brand to three major hazards. First, contamination can arise from inadequate cleanroom classification or cross‑talk between unrelated projects, compromising peptide purity and safety. Second, batch‑to‑batch variability often stems from undocumented process tweaks, leading to inconsistent assay results and wasted research funds. Finally, regulatory liability rests with the brand owner; if an FDA inspection uncovers non‑compliant practices, the brand may face warning letters, product seizures, or costly recalls.
Supplier Qualification Questionnaire
Before signing any manufacturing agreement, conduct a thorough supplier qualification questionnaire (SQQ). The SQQ should capture:
- Facility cGMP certification status and most recent audit reports.
- Details of equipment qualification, cleaning validation, and environmental monitoring programs.
- Personnel research protocols records, especially for critical operations such as peptide coupling and purification.
- Quality management system (QMS) structure, including change control and CAPA (Corrective and Preventive Action) processes.
- Availability of a Certificate of Analysis (CoA) for every released lot.
Request a sample CoA alongside the questionnaire; a well‑prepared supplier will provide a document that lists identity, purity, assay method, residual solvents, and any observed deviations.
Essential Batch Documentation
Each peptide batch must be accompanied by a complete documentation package. The core elements are:
- Manufacturing Batch Record (MBR): A step‑by‑step log of every operation, including raw‑material lot numbers, process parameters, and in‑process test results.
- Certificate of Analysis (CoA): The final analytical report confirming that the product meets predefined specifications for identity, purity, and impurity limits.
- Deviation Reports: Detailed accounts of any departures from the approved process, with root‑cause analysis and corrective actions taken.
Retaining these records for at least three years—preferably longer—demonstrates due diligence during FDA inspections and provides a clear audit trail for your researchers.
Practical Tip: Leverage a cGMP White‑Label Partner
For emerging peptide brands, partnering with a white‑label provider that already operates under cGMP can dramatically reduce the compliance burden. Such partners typically supply pre‑validated batch records, ready‑made CoAs, and a vetted supplier network, allowing you to focus on branding, packaging, and distribution. By integrating a compliant manufacturer into your supply chain from day one, you avoid costly retrofits, protect your brand’s reputation, and stay firmly within FDA expectations for Research Use Only peptides.
Compliance Checklist and Actionable Roadmap for New Brands
Step‑by‑step audit checklist
- Product classification – Confirm that every peptide you sell is labeled exclusively as “Research Use Only (RUO).” Double‑check that no language implies research-grade or dietary‑supplement use.
- Labeling requirements – Ensure each primary label includes the RUO disclaimer, the peptide’s scientific name, batch/lot number, storage conditions, and a clear statement that the product is not for human consumption.
- Documentation & record‑keeping – Maintain a master file that contains the peptide’s synthesis protocol, analytical certificates (purity, identity, sterility), safety data sheets, and a traceable chain‑of‑custody log for every batch.
- Marketing claims – Review all website copy, promotional emails, and social‑media posts. Remove any language that suggests efficacy, serving size recommendations, or clinical outcomes. Stick to factual statements about purity, research applications, and handling instructions.
- Manufacturing controls – Verify that your contract manufacturer follows cGMP‑aligned SOPs, conducts in‑process testing, and provides a full batch record. Conduct a quarterly audit or request a third‑party inspection report.
Roadmap from concept to market launch
- Idea generation (Weeks 1‑2) – Identify the peptide target, review scientific literature, and decide on the intended research niche.
- Regulatory classification (Weeks 2‑3) – Confirm RUO status, draft the RUO disclaimer, and align product naming with FDA guidance.
- Supplier selection (Weeks 3‑5) – Choose a cGMP‑compliant manufacturer, request synthesis and analytical data, and negotiate labeling services.
- Label & packaging design (Weeks 5‑6) – Create label mock‑ups that include all mandatory elements; run a compliance review with a regulatory consultant.
- Documentation assembly (Weeks 6‑8) – Compile the master file, safety data sheets, and batch records; set up a secure digital repository.
- Pre‑launch audit (Week 9) – Perform a self‑assessment using the checklist below; address any gaps before the first order is fulfilled.
- Market launch (Week 10+) – Activate your e‑commerce platform, begin dropshipping, and monitor post‑launch feedback for any compliance drift.
Quick‑fire self‑assessment questions
- Do all primary labels display the exact “Research Use Only – Not for Human Consumption” disclaimer?
- Is every batch accompanied by a Certificate of Analysis that includes purity ≥ 95 % and a full peptide identity report?
- Have you removed any serving size, research-grade, or “has been investigated for its effects on” language from product pages and marketing emails?
- Do you retain a complete chain‑of‑custody log for each batch from synthesis to final packaging?
- Has a qualified regulatory consultant reviewed your labeling, website copy, and SOPs within the past 12 months?
Ongoing compliance resources
- FDA Guidance on Research Use Only (RUO) Products
- FDA Labeling Requirements for Medical Products
- Monthly Peptide Industry Webinars (registration required)
- Professional Regulatory Consultants – Peptide Specialty
- YourPeptideBrand’s “Peptide Compliance Starter Kit” (free download)
Ready to lock down compliance before you ship your first vial? Download the free Peptide Compliance Starter Kit and get checklists, template SOPs, and a quick‑start guide that keep your brand FDA‑ready from day one.
Conclusion and Next Steps with YourPeptideBrand
Key takeaways: the six most common FDA compliance mistakes
- Misclassifying peptides as dietary supplements. Treating Research Use Only (RUO) peptides as food‑grade products invites FDA enforcement and erodes research subject trust.
- Inadequate label disclosures. Omitting required statements—such as “Not for human consumption” and batch identifiers—creates legal exposure and hampers traceability.
- Skipping the 510(k) or IND pathway when required. Assuming all peptides are exempt ignores the FDA’s risk‑based evaluation and can lead to costly recalls.
- Failing to maintain proper manufacturing records. Incomplete batch logs, SOPs, or change‑control documentation undermine Good Manufacturing Practices (GMP) compliance.
- Neglecting post‑market surveillance. Without a system to capture adverse events or quality complaints, brands cannot demonstrate ongoing safety.
- Overlooking state‑level regulations. Ignoring local pharmacy‑board requirements or specific labeling mandates can result in state‑level penalties even when federal rules are met.
Addressing each of these errors safeguards your reputation, protects research subjects, and prevents costly shutdowns. A compliant foundation not only studies have investigated effects on regulatory risk but also builds confidence among clinicians and end‑research applications who rely on the integrity of your brand.
Compliance is an ongoing commitment
The FDA does not view compliance as a one‑time checkbox. Regulations evolve, new guidance is issued, and market expectations shift. Continuous monitoring of label language, batch documentation, and post‑market data is essential. By embedding regular audits and staying abreast of guidance updates, you transform compliance from a hurdle into a competitive advantage.
Why YourPeptideBrand is the turnkey solution
YourPeptideBrand (YPB) eliminates the guesswork. Our white‑label platform delivers:
- On‑demand label printing with FDA‑approved wording.
- Custom packaging that meets both federal and state requirements.
- Direct dropshipping to clinics and researchers, removing inventory risk.
- Built‑in compliance support, from SOP creation to batch record management.
- No minimum order quantities, allowing you to scale at your own pace.
Each component is designed to keep you “ready‑to‑ship” while remaining fully aligned with FDA expectations for RUO peptide products.
Risk‑free consultation and rapid market entry
Our team of regulatory specialists has guided dozens of emerging peptide brands through the FDA maze. We offer a complimentary, no‑obligation consultation to map your specific compliance gaps and outline a clear path to market. Because we handle label generation, packaging design, and logistics, researchers may launch within weeks—not months—without worrying about minimum order constraints.
Take the next step
Ready to turn compliance into a catalyst for growth? Visit YourPeptideBrand.com to learn more and claim your free compliance starter kit. Let us handle the regulatory heavy lifting so researchers may focus on delivering high‑quality peptides under your own brand.





